

|
LINFOMAS CUTÁNEOS
Dr. José Luis Rodríguez
Peralto
Dra. Soledad Alonso García
Dpto. de Anatomía Patológica
Hospital U. 12 de Octubre
Hasta comienzos de los años 70, los únicos linfomas cutáneos bien caracterizados eran la micosis fungoide (MF), sus variantes clínicas (formas demblée y eritrodérmica), procesos relacionados (Síndrome de Sezary (SS) y reticulosis pagetoide), y la papulosis linfomatoide. El resto de los linfomas cutáneos, infrecuentes, se consideraban manifestaciones de linfomas sistémicos y se agrupaban bajo los términos de reticulosis maligna o sarcomas de células reticulares, con un pronóstico peor. En el año 1975, Edelson agrupa a la micosis fungoide con sus variantes y al síndrome de Sezary bajo el epónimo de Linfoma cutáneo de células T. El término tiene mucha aceptación, y grandes desventajas, como no diferenciar MF, SS y otros linfomas cutáneos T de clínica y evolución diferentes. A final de los años 70 se introduce la inmunohistoquímica que permite distinguir dos grandes categorías, linfomas cutáneos B y T. En Europa, se utiliza la clasificación de Kiel para subclasificar los linfomas B cutáneos de la misma forma que se hacía en los gánglios linfáticos. En América, se adoptó la Working Formulation, que no establecía diferencias entre linfomas B y T. Pronto se advierte que estas clasificaciones eran poco útiles aplicadas a la piel. En el año 1994, se realiza una clasificación Internacional de consenso de linfomas, fruto de la cual surge la clasificación REAL que trata de distinguir diferentes categorías, relacionando por primera vez sus aspectos morfológicos, inmunohistoquímicos y genéticos con los evolutivos clínicos. En el año 1997, Willenze y un grupo danés, separan claramente los linfomas cutáneos primarios de los secundarios y establecen la clasificacion EORTC de linfomas cutáneos primarios. Consideran linfoma cutáneo primario, aquél que en el plazo de 6 meses tras la aparición de su lesión en piel, no presenta afectación extracutánea mediante los métodos de imagen (TAC corporal) y tras la realización de médula ósea. Los linfomas que afectan secundariamente o concurrentemente la piel, los clasificaremos según el diagnóstico del linfoma inicial.
Nosotros, actualmente tratamos de clasificar los linfomas cutáneos primarios según la clasificación REAL, pero considerando las siguientes premisas:
a) Distinguir inicialmente entre linfoma cutáneo primario y secundario, basado en la ausencia de tumor extracutáneo en un plazo de 6 meses desde el diagnóstico del linfoma en piel.
b) Realizar el diagnóstico correlacionando siempre clínica, histología e inmunohistoquímica. Si no aunamos estos tres datos, el diagnóstico final no será nunca correcto.
Basándonos en estos criterios, distinguimos las siguiente categorias de linfomas cutáneos primarios:
LINFOMAS T:
·
Micosis fungoide
y variantes
·
Papulosis Linfomatoide
·
Linfoma T de células
grandes CD 30+
·
Linfoma T periférico
LINFOMAS B:
·
Linfoma de la zona
marginal
·
Linfoma de células
grandes
·
Linfoma Folicular
·
Plasmocitoma
LINFOMAS CUTÁNEOS INFRECUENTES:
·
Linfoma intravascular
B ó T
·
Linfoma T paniculítico
·
Linfoma NK blástico
LINFOMAS T CUTÁNEOS PRIMARIOS
El problema de diagnóstico
de los linfomas T cutáneos
es quizá más simple que el de los linfomas B, puesto que la mayoría de
los cuadros están perfectamente delimitados. La gran mayoría de los casos
corresponden a Micosis Fungoide (MF) o alguna de sus variantes (mucinosis folicular,
síndrome de la piel laxa, síndrome de Sezary, etc), seguida en frecuencia
de la Papulosis Linfomatoide.
El resto de los tumores son prácticamente proliferaciones difusas de células
grandes que inmunohistoquímicamente
pueden expresar el antígeno de superficie CD30 o no; correspondiendo a
linfomas anaplásicos de células grandes CD30+ o a linfomas T periféricos,
respectivamente. Quizá el problema más importante resida en diferenciar
los linfomas T CD30+ de la papulosis linfomatoide, basándose su diagnóstico
diferencial en el aspecto clínico de las lesiones (nódulos-tumores o pápulas).
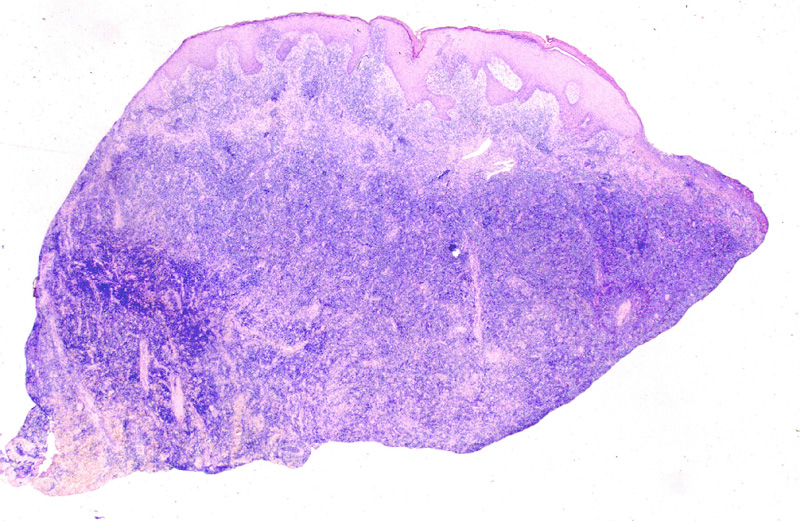
MICOSIS FUNGOIDE.-
La Micosis Fungoide (MF) es el linfoma primario por excelencia de la
piel. Se caracteriza por una proliferación de linfocitos T neoplásicos
epidermotropos, de tamaño pequeño o mediano con núcleos cerebriformes.
Suelen tener un curso en general indolente con progresión lenta de años
o décadas. Así, evolucionan desde la fase en placa, a placa infiltrada
(Fig.1) y tumor (Fig.2).
En estadios avanzados los tumores suelen ser muy agresivos, con dos formas
histológicas, aquellos constituidos por células pequeñas cerebriformes
y los de aspecto anaplásico con células grandes que incluso pueden expresar
el antígeno CD 30.
{kind=link}
{kind=link}
Microscópicamente, la MF en
estadios iniciales puede presentar cualquiera de los patrones dermatopatológicos
conocidos, siendo el de dermatitis psoriasiforme y liquenoide los más
frecuentes y el de dermatitis espongiforme uno de los más raros, de tal
forma que, ante la presencia de clara espongiosis se debe descartar previamente
un eczema antes de pensar en micosis fungoide.
Un patrón dermatopatológico muy sugestivo de MF es el de Mucinosis folicular. Así, ante la presencia
de mucinosis en los folículos, lo primero que debemos plantearnos es una MF o un linfoma T cutáneo, y una vez descartado
histológicamente, realizar reordenamiento de receptores T, puesto que
muchos de estos cuadros reordenan monoclonalmente y otros evolucionan
con el tiempo a un linfoma cutáneo T.
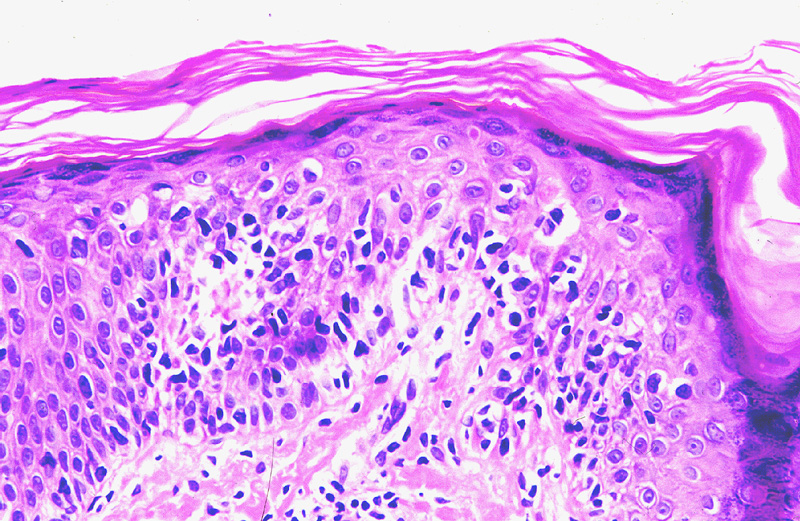
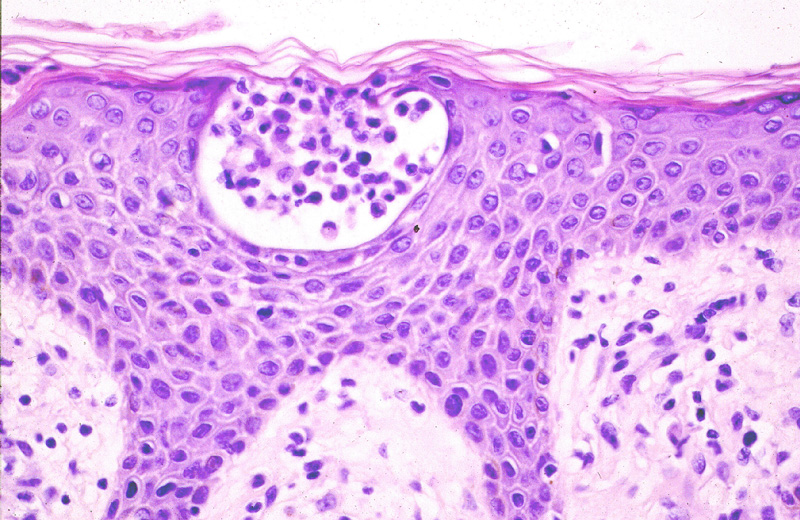
Los criterios microscópicos
típicos diagnósticos de la micosis fungoide, especialmente en los estadios
iniciales, son los clásicos recogidos por Smoller: Linfocitos con halo, exocitosis linfocitaria desproporcionada, linfocitos
alineados en la basal (en fila india) (Fig.3),
microabscesos de Pautrier (Fig.4), hiperlobulación
de los linfocitos epidérmicos y linfocitos epidérmicos mayores que los
dérmicos. Ackerman, además de todos estos datos, ha destacado la presencia
en la dermis de un infiltrado liquenoide asociado a haces de colágeno
muy finos (colágeno en alambre). De todos estos datos, el más constante
es la presencia de halo claro perinuclear
en linfocitos hipercromáticos hiperlobulados intraepidérmicos y el
más infrecuente la presencia de nidos de Darier, también conocidos como
microabscesos de Pautrier.
{kind=link}
{kind=link}
Se han descrito múltiples variantes
clínicas e histopatológicas de micosis fungoide. Las más significativas
son: MF ampollosa, MF tipo acantosis nigricans, MF dishidrótica, MF eritrodérmica,
mucinosis folicular, MF mucinosa, MF folicular, MF granulomatosa,
slack skin syndrome (forma granulomatosa con destrucción de fibras
elásticas), MF palmoplantar, reticulosis pagetoide, MF poiquilodérmica,
MF pustulosa, MF hiperqueratótica/pustulosa, MF ictiosiforme, MF hipopigmentada,
MF con quistes eruptivos, MF hiperpigmentada, MF similar a la dermatitis
peioral, MF tipo púrpura pigmentada, MF zosteriforme, MF angiocéntrica.
El diagnóstico diferencial
es muy amplio, pero uno de los problemas más importantes es diferenciar MF de eccema, con el que clínicamente presenta
grandes similitudes pero que histológicamente se puede diferenciar si
atendemos a la ausencia de espongiosis en la epidermis de la MF, hecho
constante en el eczema. La espongiosis con incluso formación de vesículas
es un hecho que por sí mismo va en contra del diagnóstico de MF, aunque
no podemos olvidar que existe una variedad de MF espongiótica, muy rara,
cuyo diagnóstico está basado en la presencia de linfocitos intraepidérmicos
atípicos con halo perinuclear.
PAPULOSIS LINFOMATOIDE.-
La
Papulosis linfomatoide es un linfoma T
cutáneo que se caracteriza clínicamente por la presencia de múltiples
pápulas o pequeños nódulos generalmente en piel de tronco y región proximal
de extremidades que regresan y recidivan espontaneamente durante años.
Suelen afectar a adultos jóvenes, aunque también se han descrito en niños.
A veces se asocian a otros tipos de linfomas cutáneos siendo la MF el
más frecuente. Las pápulas suelen ser rosadas o marrones con frecuente
ulceración central.
Microscópicamente,
se reconocen tres variedades perfectamente diferenciables histológicamente,
pero sin implicación pronóstica (tipos A,
B y C). Estas pueden aparecer de forma sincrónica o en distintos
momentos de la evolución de un mismo paciente. El dermatopatólogo debe
saber identificarlas para conocer las distintas caras de esta enfermedad.
Tipo A (Tipo histiocítico): Es la más
frecuente, se caracteriza por una proliferación polimorfa de linfocitos,
histiocitos, neutrófilos y eosinófilos mezclados con células atípicas
aisladas o en pequeños grupos que ocupan la dermis papilar y reticular,
adquiriendo una forma triangular con base hacia la epidermis, que con
frecuencia se encuentra necrosada y ulcerada (Fig
5).
{kind=link}
Tipo B (Tipo linfocítico o similar a la Micosis
Fungoide): Es una forma rara, que se caracteriza por una proliferación
en banda de linfocitos atípicos, pequeños, hipercromáticos y cerebriformes,
con epidermotropismo. Esta forma, sólo desde el punto de vista
histológico, es indistinguible de una micosis fungoide.
Tipo C (Tipo linfoma de células grandes):
Se trata de una proliferación linfoide en sábana de células grandes (Fig.6)
acompañadas de escasos linfocitos, neutrófilos y eosinófilos. La única
forma de distinguirlo de un linfoma T de células grandes CD30 + cutáneo es por la clínica, presencia sólo de pápulas y ausencia
de nódulos o tumores cutáneos.
{kind=link}
Las tres variedades suelen
expresar marcadores T helper es decir, son CD3+, CD43+, CD4+ y CD8-.
En algunas ocasiones se pueden perder algunos antígenos pan T. En cuanto
a las células grandes son CD30+ en los tipos A y C. En el tipo B, no suele
existir expresión de CD30. La positividad del CD30 debe ser citoplasmática
y paranuclear (en el aparato de Golgi) en células aisladas o formando
pequeños grupos, dato que facilita el diagnóstico diferencial con procesos
inflamatorios.
LINFOMA T DE
CÉLULAS GRANDES CD30+.
Este
linfoma es similar histopatológicamente a la papulosis linfomatoide tipo
C con la salvedad de que clínicamente no se manifiesta como pápulas sino
como nódulos grandes, ulcerados, de color rojo-marrón, que a veces regresan
espontáneamente.
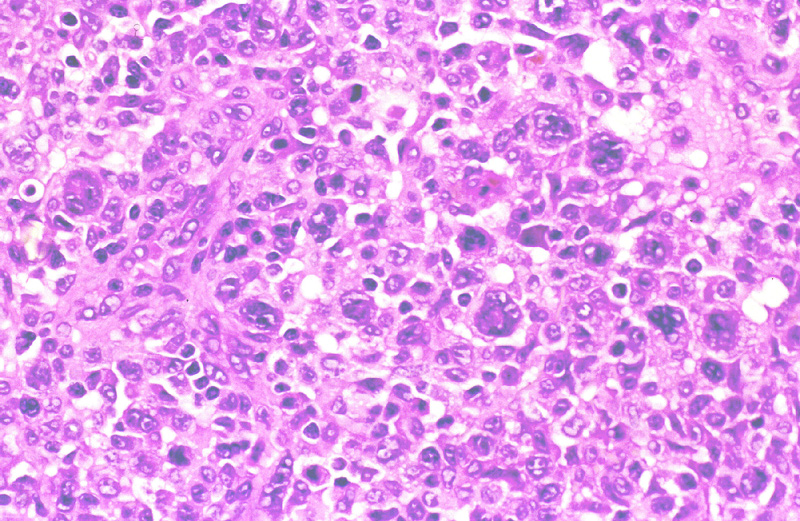
Histológicamente, se presenta como una proliferación difusa o nodular
de linfocitos grandes, atípicos, de citoplasma evidente y nucleolo prominente
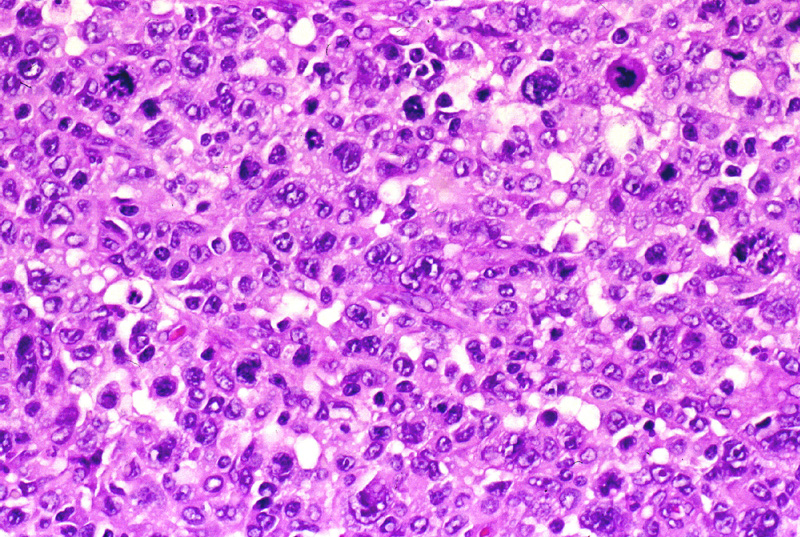
que puede ser indistinguible de la papulosis linfomatoide tipo C (Fig.7),
especialmente en los casos ulcerados, con hiperplasia epidérmica e infiltrado
inflamatorio. Su fenotipo inmunohistoquímico es T: CD3 +, CD43 + y CD4
+, con expresión membranosa y paranuclear de CD30 (Fig.8).
No expresan ALK ni muestran la traslocación t (2;5).
{kind=link}
{kind=link}
Aunque microscópicamente es un linfoma de alto grado su evolución
es generalmente favorable. Es infrecuente que maten al paciente. Las lesiones
cutáneas aisladas se tratan con radioterapia local mientras que las generalizadas
o con afectación ganglionar con quimioterapia.
LINFOMA T PERIFÉRICO.
Este tumor ha recibido diversas denominaciones
tales como: linfoma T inmunoblástico, linfoma T pleomórfico o la actualmente
aceptada de linfoma T periférico. Son linfomas raros, agresivos y de evolución
generalmente fatal.
Clínicamente se presentan como
tumores o placas grandes a menudo ulceradas. Para su diagnóstico es imprescindible
descartar clínicamente una micosis fungoide previa con transformación
tumoral.
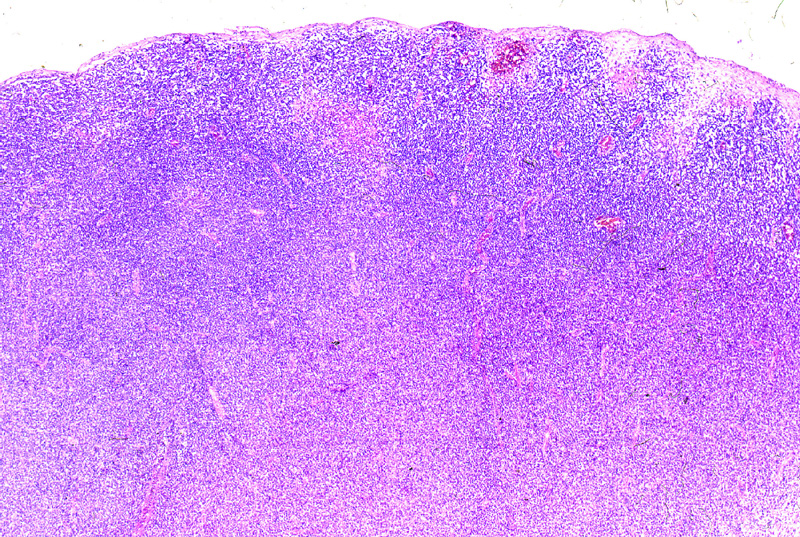
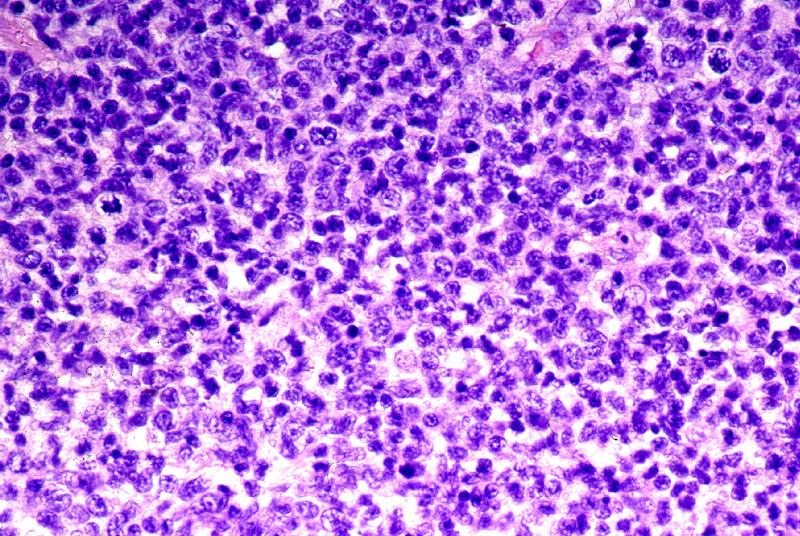
Microscópicamente, aparecen
como una infiltración difusa o nodular de toda la dermis y tejido celular
subcutáneo (Fig.9) caracterizada por células
grandes o de mediano tamaño, de citoplasmas amplios y núcleos grandes,
irregulares, a veces con nucleolo evidente (Fig.10).
No suelen presentar epidermotropismo, siendo el índice mitósico muy elevado.
Inmunohistoquímicamente, su fenotipo es T: CD3+, CD43+ y CD4+, con
negatividad para el CD30 -.
{kind=link}
{kind=link}
El tratamiento de elección
es quimioterapia sistémica.
LINFOMAS B CUTÁNEOS PRIMARIOS
Los linfomas B cutáneos primarios
son más infrecuentes que los linfomas T, aunque aparecen con mayor frecuencia
de lo que se piensa. Gracias al uso de las técnicas moleculares e inmunohistoquímicas,
se ha demostrado que muchos de los pseudolinfomas diagnosticados antaño
son en realidad linfomas B. Hasta los estudios de la EORTC impulsados
por Willenze no estaban bien definidos los criterios diagnósticos del
linfoma B primario. Actualmente, la aparición de la clasificación REAL
y de la OMS y su aplicación a la piel, ha servido para clarificar y simplificar
su clasificación.
LINFOMA DE LA ZONA MARGINAL.
Es el linfoma B primario de piel más
frecuente. Muchos de estos tumores están incluídos en nuestros archivos
como pseudolinfomas o linfocitomas cutis y en otras series como linfomas
foliculares o inmunocitomas. Este último término preferimos eliminarlo
e incluirlo dentro de este epígrafe.
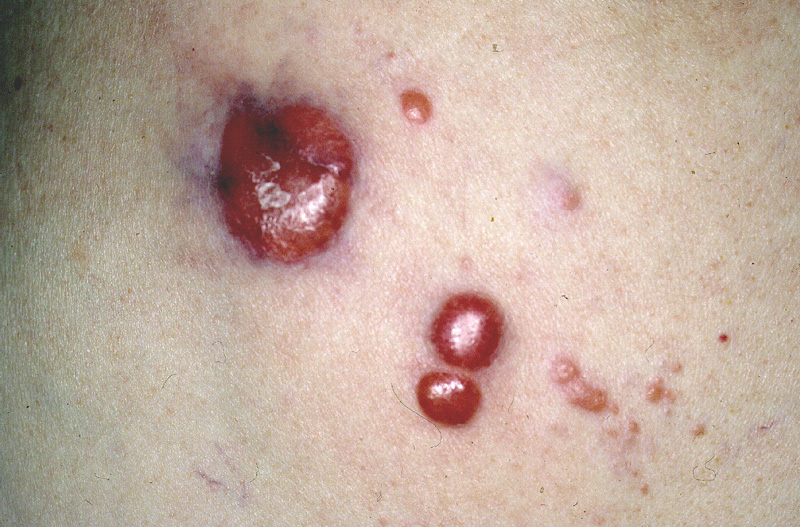
Clínicamente, se presentan como nódulos,
placas o pápulas múltiples (Fig.11), recidivantes,
preferentemente en extremidades superiores o tronco. El pronóstico es
excelente, siendo excepcional que causen la muerte del paciente.
{kind=link}
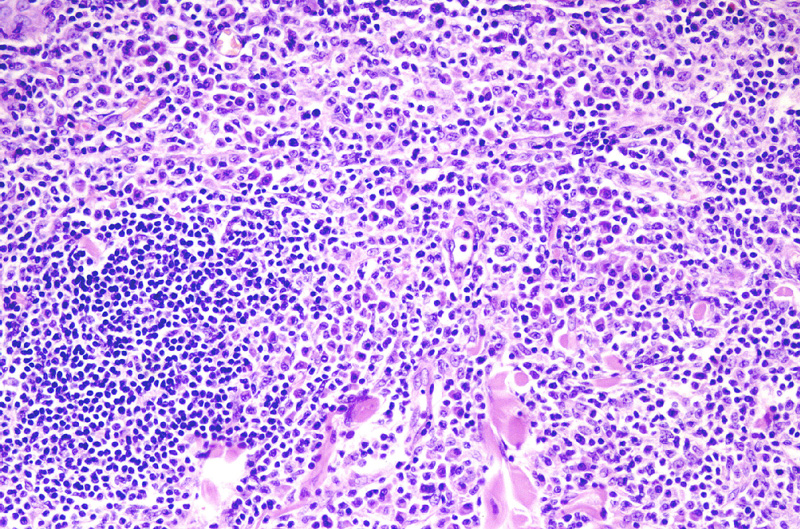
Microscópicamente, se caracteriza por
presentar una ocupación parcheada, multinodular (Fig.12)
o difusa de la dermis reticular, sin extensión a la epidermis. Con el
pequeño aumento, se advierten frecuentes folículos linfoides reactivos
constituídos por centros germinales bien estructurados con una zona de
manto periférica, rodeados por una proliferación de células polimorfas,
entre las que destacan células pequeñas hendidas tipo centrocito, grandes
blásticas con nucleolo evidente sin constituir grandes nidos y células
pequeñas de citoplasma claro y hábito monocitoide (Fig.13).
Además, es muy frecuente la presencia de grupos de células plasmáticas
o linfoplasmocitoides.
{kind=link}
{kind=link}
Inmunohistoquímicamente, las células
pequeñas, tipo centrocito o monocitoides y las grandes blásticas muestran
fenotipo B: CD20+, y CD79a+, que se
suelen acompañar de abundantes células pequeñas T reactivas, por lo que,
en algunas ocasiones es muy útil para diferenciarlo de picaduras de artropodo
ver si existe restricción de cadenas ligeras Kappa o Lambda. El tratamiento
es excisión quirúrgica en las lesiones solitarias
y radioterapia local. El uso de corticoides sistémicos también
ha demostrado su eficacia. La quimioterapia sólo se utiliza para aquellos
casos excepcionales con afectación extensa o difusa.
LINFOMA DE CÉLULAS GRANDES.
Son linfomas cutáneos primarios, que
clínicamente aparecen en adultos de alrederor de 50 años (22-88 a.) como
lesiones frecuentemente tumorales, únicas
o múltiples, que raramente desaparecen espontáneamente. En un 40% de los
pacientes pueden aparecer también placas y pápulas. La evolución suele
ser buena, aunque recidivan tras el tratamiento en un 34% de los casos, a veces con extensión ganglionar, pero es excepcional
que causen la muerte del paciente (3%).
Microscópicamente, se caracterizan
por una proliferación difusa o nodular confluente que ocupa masivamente
la dermis reticular, infiltrando el tejido adiposo subyacente (Fig.14).
El tumor está predominantemente constituido por células linfoides de gran
tamaño (Fig.15), que a veces se acompañan de
células pequeñas, entremezcladas o formando un ribete periférico. En estos
casos, se pueden identificar folículos linfoides. Inmunohistoquímicamente,
el tumor expresa fenotipo B: CD20+, CD79a+, siendo Bcl2 + en sólo un 28%,
bcl6 + en un 41% y CD10 + en un 37%.
{kind=link}
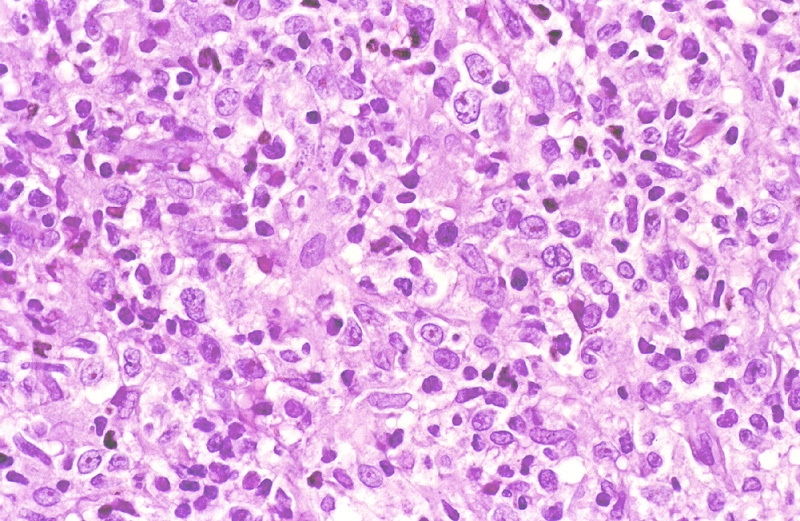{kind=link}
El tratamiento de elección es la radioterapia
local y poliquimioterapia en los casos con extensión sistémica.
LINFOMA FOLICULAR.
Aunque en algunas series constituye
el mayor grupo de linfomas B primario cutáneo, nosotros consideramos que
se trata de un tumor raro en la piel especialmente si se es estricto a la hora de realizar su diagnóstico. Clínicamente,
aparecen en adultos de edades comprendidas entre 22 y 72 años como placas
o tumores y a menudo nódulos únicos. Recidivan en un 44% de los casos,
a veces con extensión ganglionar, pero en ninguno de los 18 casos revisados
ha llevado a la muerte del paciente.
El diagnóstico histopatológico requiere
ser muy estricto. Se caracteriza por una ocupación homogenea de la dermis
en forma de nódulos, a menudo confluentes, que ocasionalmente se extienden
al tejido adiposo. Además de esta arquitectura nodular, es imprescindible
la presencia de folículos tumorales en el centro de la lesión (Fig.16).
La epidermis suele estar respetada Grenz zone. Los folículos tumorales
están compuestos por clásicos centrocitos y centroblastos en diferentes
proporciones, con un manto mal definido y ausencia de macrófagos en patrón
de cielo estrellado. De forma ocasional se reconocen folículos linfoides
reactivos en la periferia. Inmunohistoquímicamente, presentan un fenotipo
CD20+, CD79a+, bcl6+ en todos los casos, CD10+
en el 87%, y bcl2+ en el 61%. La mayoría de los casos presentan
también expresión de bcl6 ó CD10 en las áreas interfoliculares. En el
43% de los casos se observa restricción de cadenas ligeras (Kappa habitualmente).
En el centro de los folículos se identifican células dendríticas CD23
ó CD21 positivas. Es frecuente la presencia de células reactivas T, CD3
+. No se identifica la traslocación t(14;18) en estos tumores.
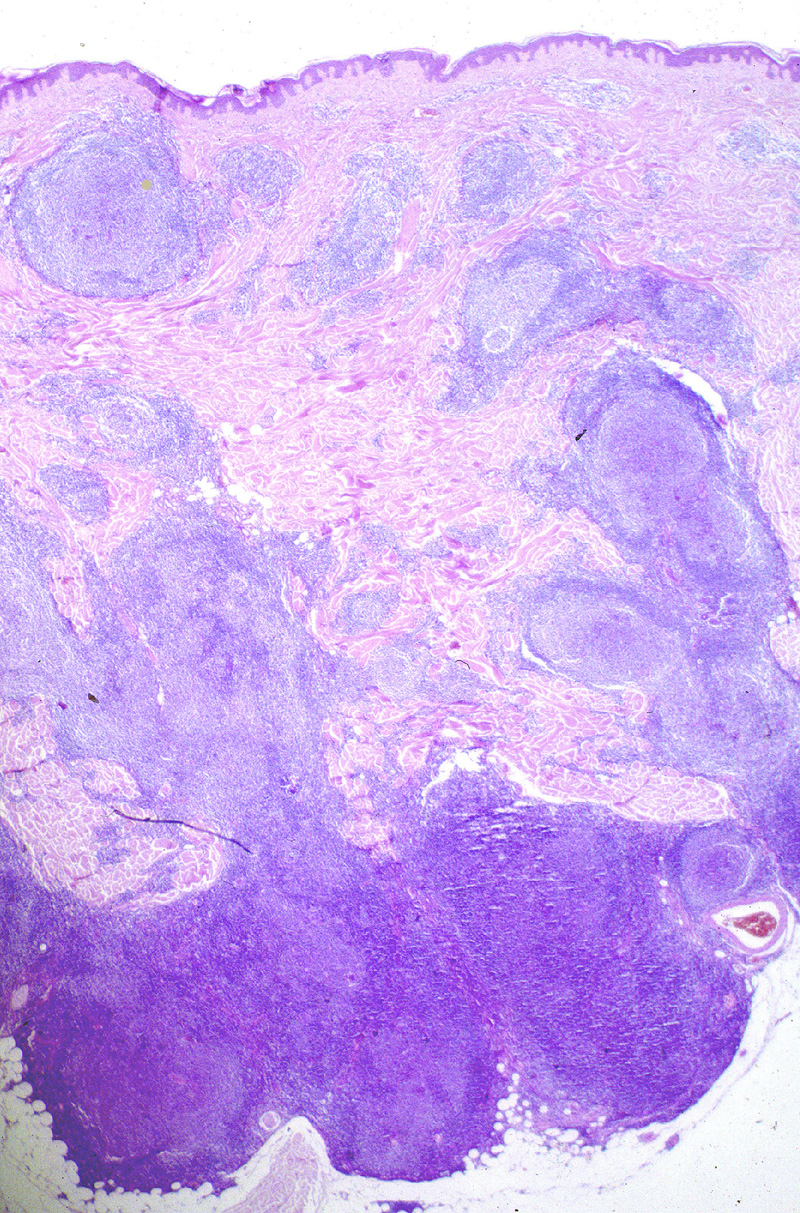{kind=link}
El
tratamiento consiste en radioterapia local.
PLASMOCITOMA.
Es un tumor extremadamente raro, que
se caracteriza por una proliferación monoclonal de células plasmáticas localizadas exclusivamente en la piel, en ausencia
de afectación de médula ósea. Clínicamente, se caracteriza por la presencia
de nódulos únicos o múltiples, o excepcionalmente placas eritematosas,
infiltradas, de localización preferente en cabeza y tronco, en pacientes
de edad avanzada. El pronóstico de estos tumores es mucho mejor que el
de los pacientes con afectación cutánea secundaria por plasmocitoma múltiple.
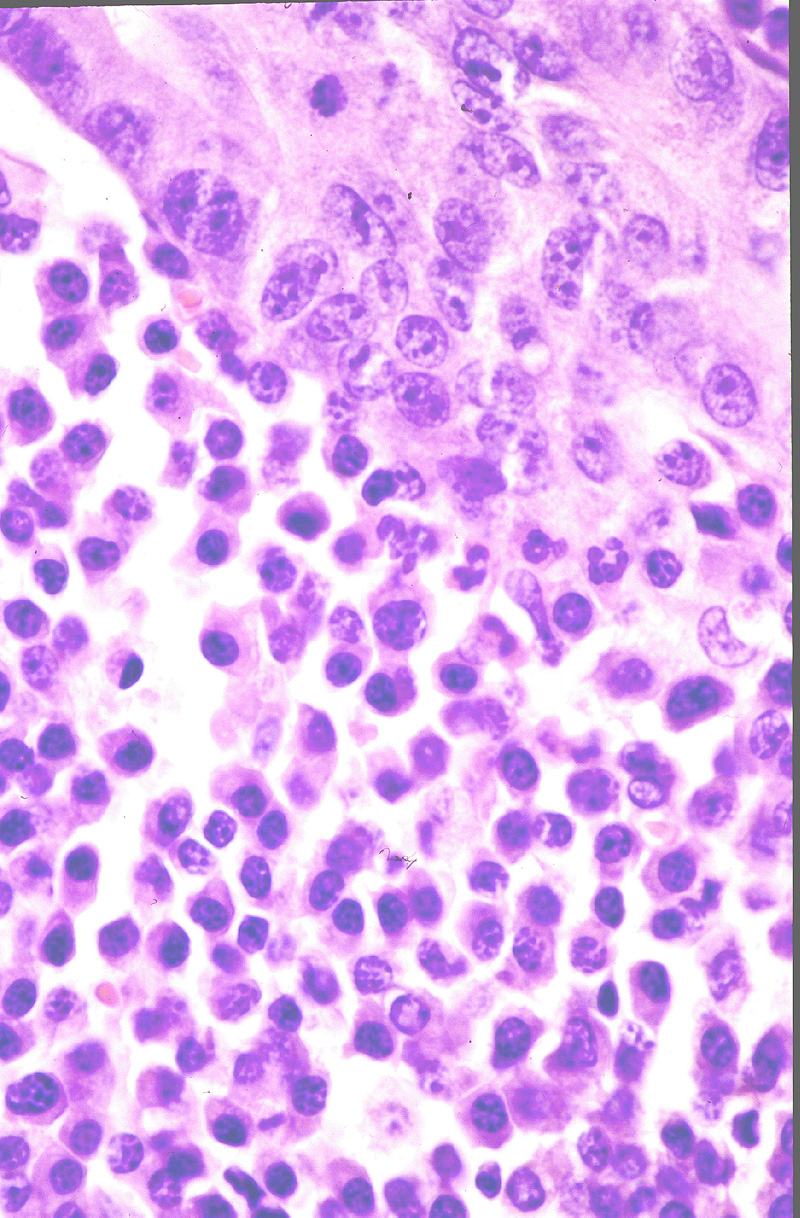
Microscópicamente, se caracteriza por
una proliferación nodular (Fig.17) o difusa
que ocupa masivamente la dermis y subcutis. El tumor está constituido
casi exclusivamente por células plasmáticas maduras e inmaduras con un
grado de atipia variable (Fig.18). Se observan
con frecuencia Cuerpos de Dutcher y Russell. El infiltrado linfoide reactivo
es escaso o ausente. Aunque se ha descrito amiloide dentro o rodeando
la neoplasia, su presencia es más frecuente en aquellos casos de afectación
secundaria cutánea por plasmocitoma. Inmunohistoquímicamente, se caracteriza
por presentar un fenotipo CD38+, CD138+, y por contener un tipo de Ig,
habitualmente IgA. La negatividad para CD45 y la expresión de HMB45, Citoqueratinas
y CD30, puede presentar problemas de diagnóstico diferencial.
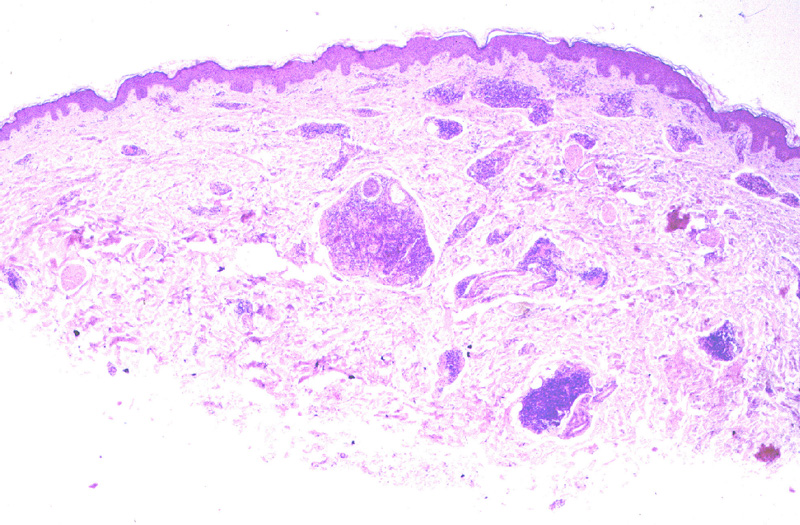{kind=link}
{kind=link}
El tratamiento de elección es radioterapia
local o cirugía.
LINFOMAS CUTÁNEOS INFRECUENTES.
Bajo este epígrafe hemos incluído todos
aquellos linfomas primarios cutáneos extremadamente raros, algunos de
los cuales no son encuadrables dentro de las categorías de linfomas B
ó T, bien porque puedan expresar ambos fenotipos o porque no expresen
ninguno de ellos.
LINFOMA INTRAVASCULAR B ó T.
Se trata de proliferaciones linfoides
monomorfas de células grandes intravasculares. Habitualmente expresan
marcadores B, sin embargo se han descrito algunos casos T. Estos tumores
se han considerado durante muchos años neoplasias vasculares, denominandose
angioendoteliomatosis maligna.
Clínicamente suelen afectar la piel
y el sistema nerviosos central, aunque existen algunos casos con infiltración
exclusiva cutánea. Los pacientes presentan placas induradas eritematosas
o violaceas en tronco y muslos, sugestivas inicialmente de paniculitis.
La evolución es generalmente mala.
Microscópicamente se observa una proliferación
de linfocitos grandes que rellenan masivamente los vasos sanguineos de
la dermis y el tejido celular subcutáneo, con extensión focal extravascular.
Las células son grandes, de citoplasma escaso y núcleos grandes con nucleolo
evidente. Inmunohistoquímicamente, las células expresan fenotipo B: CD20+
y CD79a+ en la mayoría de los casos, aunque se han descrito algunos casos
raros con fenotipo T: CD3+ y CD5+.
El
tratamiento consiste en poliquimioterapia sistémica.
LINFOMA T PANICULÍTICO.
En la literatura existe gran confusión con este tumor, puesto que se ha
hecho sinónimo a la paniculitis histiocítico-citofágica. Actualmente,
se considera que el espectro paniculitis histiocítica-citofágica incluye
dos tipos diferentes de lesiones. Por un lado, aquellas secundarias a
procesos autoinmunes, en los que sólo se advierte una paniculitis lobulillar
con eritrofagocitosis (de buen pronóstico) y un segundo grupo, al que
nos vamos a referir a continuación, que consiste en un linfoma T paniculítico
que se acompaña de eritrofagocitosis.
Los linfomas T paniculíticos se caracterizan
clínicamente, por presentarse en pacientes adultos como tumores o placas
infiltradas solitarias o múltiples, generalmente no ulceradas, en las
extremidades y menos frecuentemente en tronco y cabeza. Las lesiones pueden
simular clinicamente eritema nodoso, aunque se acompañan de fiebre, malestar
general, cansancio, y pérdida de peso. Es frecuente un cuadro hemofagocítico
en los estadios avanzados. El pronóstico es fatal, conduciendo rápidamente
a la muerte.
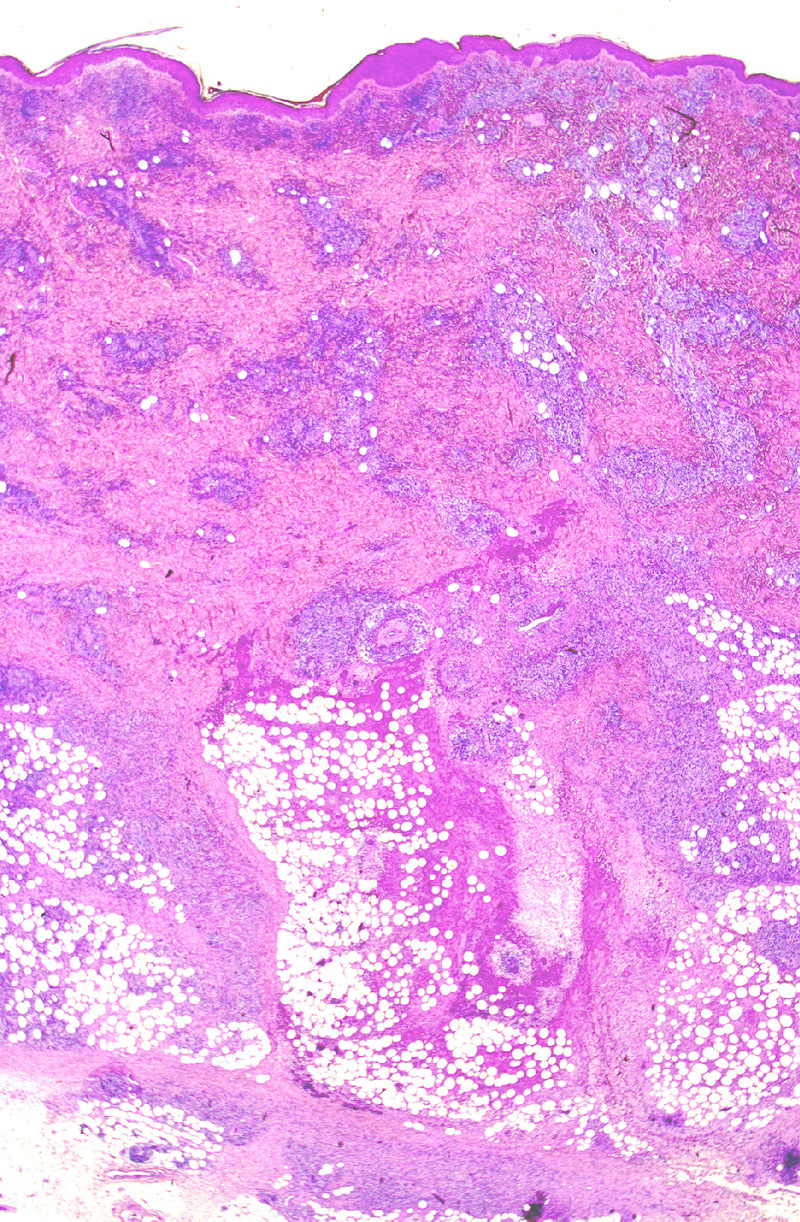
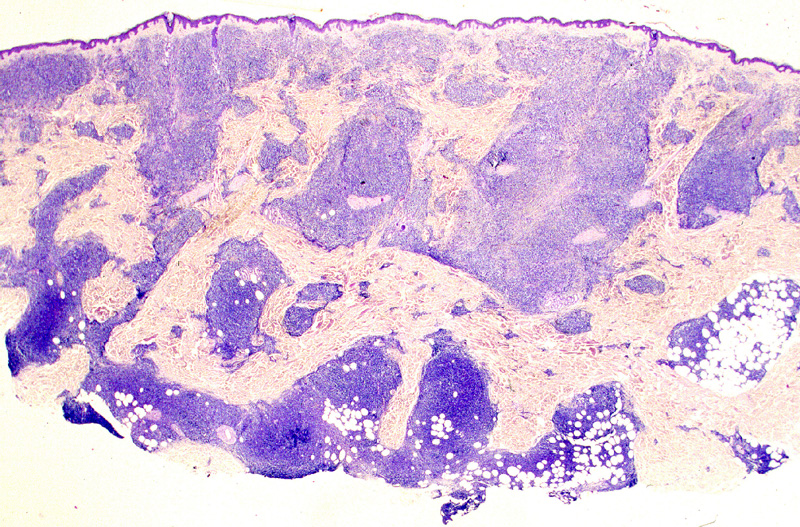
Microscópicamente, se caracteriza por
presentar una ocupación difusa o nodular del lobulillo adiposo (Fig.19),
con necrosis grasa (Fig.20) que simula una
paniclulitis lobulillar. En la dermis reticular profunda
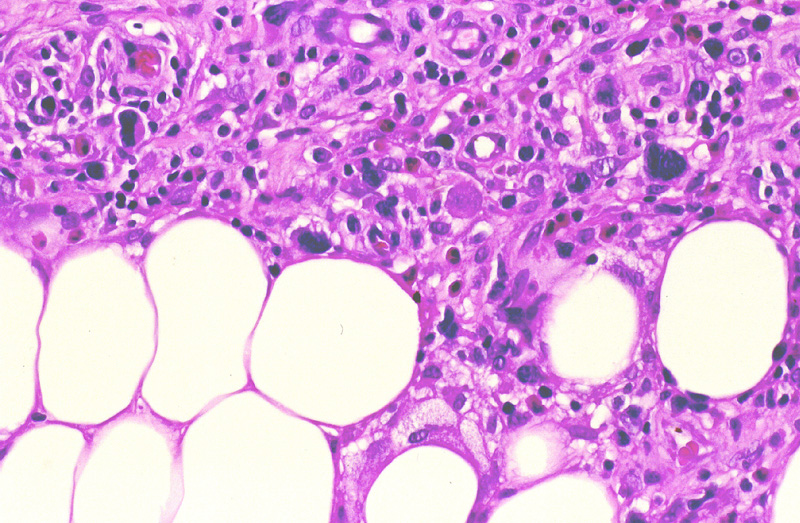
se suele reconocer nidos linfoides con infiltración perivascular
(Fig.20). Las células linfoides son pleomórficas
grandes, medianas y a veces pequeñas (Fig.21).
Es frecuente la presencia de hemofagocitosis caracterizada por grandes
macrófagos que fagocitan linfocitos neoplásicos y eritrocitos adquiriendo una morfología como en saco de habichuelas (Fig.22).
Inmunohistoquímicamente, las células linfoides muestran un fenotipo T:
CD3+, CD43+, CD4+ ó CD8+. El CD56 es negativo.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
El tratamiento de elección es quimioterapia
sistémica.
LINFOMA NK BLÁSTICO.
Se caracteriza clínicamente por aparecer
en adultos como lesiones únicas o múltiples, nodulares o en forma de placas
tumorales induradas. Además, en el momento del diagnóstico, en un porcentaje
elevado de casos, suelen tener afectación de médula ósea con expresión
en sangre periférica en forma de leucemia, por lo que muchos autores prefieren
actualmente denominarlos linfomas hematocutáneos. Existen muy pocos casos
publicados en la literatura y todos ellos con un pronóstico fatal, respondiendo
inicialmente al tratamiento con remisión completa, pero volviendo a recaer
de nuevo, con extensión visceral y muerte del paciente en poco tiempo.
Miscroscópicamente, se caracteriza
por una ocupación difusa o multinodular confluente de la dermis reticular
y tejido celular subcutáneo (Fig.23) por linfocitos
pleomórficos de tamaño grande o mediano (Fig.24)
con elevado índice mitósico. Inmunohistoquímicamente, se observa
negatividad para marcadores B y algunos T: CD20 -,
CD79a , CD3 y positividad para CD43 +, CD4 +, CD56 + (Fig.25) y CD123 +. El CD5 +/- y CD57 -. Por ello, ante un linfoma cutáneo
que expresa CD43, pero no CD3, siempre debemos ampliar la batería diagnóstica
inmunohistoquímica y comprobar si expresa CD56 y CD4, para evitar diagnosticarlos
equivocadamente como linfomas T primarios cutáneos, con las consiguientes
repercusiones (graves) para el paciente.
{kind=link}
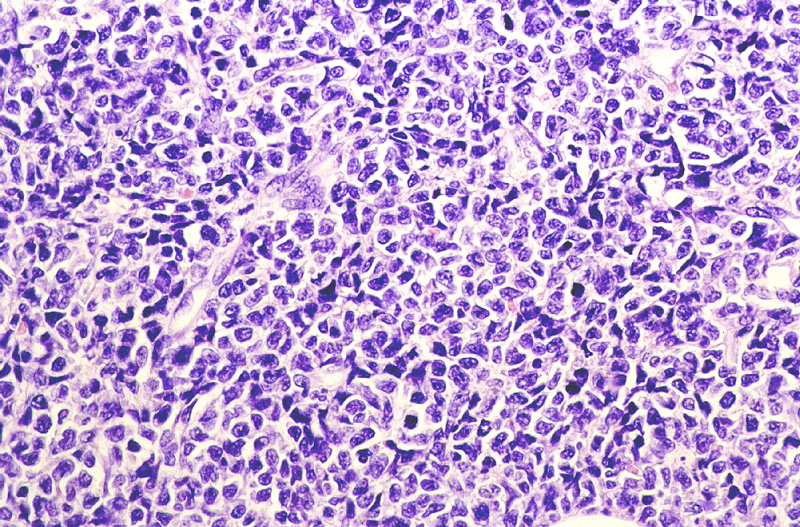{kind=link}
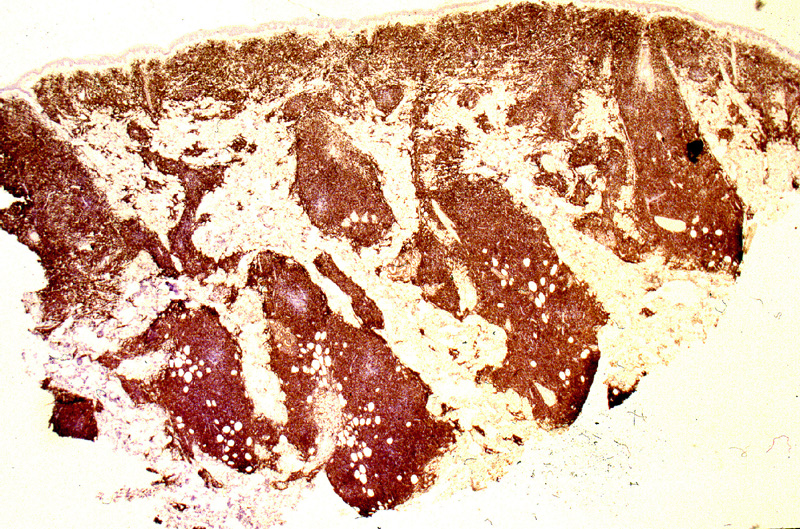{kind=link}
El tratamiento que se ha propuesto hasta ahora es la quimioterapia sistémica, con malos resultados.
PIES DE FOTO
Figura 1.- Imagen panorámica de Micosis Fungoide
(MF) en estadio de placa infiltrada.
Figura 2.- Imagen panorámica de Micosis Fungoide
(MF) en estadio tumoral.
Figura 4.- Micosis Fungoide (MF) con microabscesos
de Pautrier.
Figura 7.- Linfoma T de células grande CD 30 +.
Figura 8.- Intensa positividad para CD 30 (membranosa
y paranuclear) en el linfoma de la figura 7.
Figura 9.- Imagen panorámica de un linfoma T periférico.
Figura 11.- Imagen clínica de un paciente con
linfoma primario cutáneo de la zona marginal.
Figura 14.- Imagen panorámica de linfoma B de
células grandes.
Figura 17.- Imagen panorámica de plasmocitoma
cutáneo. Nótese la distribución multinodular.
Figura 19.- Linfoma T paniculítico. La imagen
panorámica simula una paniculitis lobulillar.
Figura 23.- Linfoma
NK blástico. Imagen panorámica.
Figura 24.- Detalle de la figura 23. Las células
atípicas son de mediano tamaño, con atipia marcada.
![]() Conganat-
2002
Conganat-
2002
Uninet.edu & Club de Informática Aplicada de la SEAP
Correo-e contacto mailto:conganat@uninet.edu