

|
GRANULOMAS EN EL PULMON. ENFOQUE DIAGNOSTICO.
Dr.Jesús Allende, Sección de Neumología,
Dra. Inmaculada Herráez, Servicio de Radiodiagnóstico.
Dra. Nieves Alonso, Servicio de Anatomía Patológica.
Hospital de León, León
GRANULOMAS
EN EL PULMON. ENFOQUE DIAGNOSTICO.-
Dr. Jesús Allende, Sección de
Neumología
Hospital de León, León
El hallazgo de granulomas en una biopsia procedente de aparato
respiratorio constituye un problema que se da con cierta frecuencia en
la práctica clínica. En ocasiones el diagnóstico resulta fácil, como cuando
se obtienen granulomas en la biopsia transbronquial de un paciente joven
con eritema nodoso y adenopatías hiliares bilaterales y paratraqueales
derechas. Pero en otros casos el diagnóstico de certeza no es tan obvio.
En cualquier situación resulta imprescindible que se produzca una estrecha
colaboración entre el patólogo, el radiólogo y el clínico.
El enfoque diagnóstico de una enfermedad granulomatosa pulmonar
obliga a tomar en consideración los datos ofrecidos por la historia clínica,
exploración física, métodos de diagnóstico por imagen (radiología, TAC
y gammagrafía con galio), exploración funcional respiratoria y hallazgos
de laboratorio. Un adecuado análisis de todos ellos nos permitirá determinar
si es preciso o no tomar una biopsia para llegar a un diagnóstico de certeza
y, en caso afirmativo, cuál es el lugar más adecuado para ello.
Desde un punto de vista práctico, la mayor parte de las causas
de granulomas en el pulmón se pueden determinar contestando sucesivamente
a las siguientes preguntas:
1.
¿ Son de causa infecciosa
?
2.
¿ Existe algún agente externo
causal ?
3.
¿ Son granulomas de cuerpo
extraño ?
4.
¿ No existe causa externa
aparente ?
Ciertas infecciones como la tuberculosis, enfermedades por micobacterias no
tuberculosas, algunas micosis y la neumonía por P carinii pueden producir reacción granulomatosa pulmonar. El diagnóstico
en este caso se realiza teniendo en cuenta el cuadro clínico-radiológico
y los hallazgos microbiológicos. Es muy importante conocer si el paciente
tiene algún factor que condicione inmunosupresión.
En otras ocasiones la historia
clínica revela la presencia de algún agente
externo capaz de producir granulomas. Así ocurre con:
+ Neumoconiosis
+ Alveolitis alérgicas extrínsecas
+ Fármacos:
Dilantina
Metotrexate
BCG
Sustancias
lipídicas
Oxifenilbutazona
Cromoglicato
disódico
Antiretrovirales
+ Sustancias diversas
Berilio
Metal duro
Nebulizadores
de laca
Una situación especial la constituyen
los llamados granulomas de cuerpo
extraño, como ocurre en pacientes sometidos a diálisis (por celulosa),
usuarios de drogas por vía parenteral (en relación con talco), aspiración
de alimentos, inhalación de diversas sustancias, microlitiasis alveolar
y amiloidosis nodular.
Cuando no se encuentra causa aparente de la presencia de granulomas en el
pulmón hay que tener en cuenta los hallazgos de:
+ Laboratorio: la presencia de
eosinofilia apunta a granulomatosis alérgica de Churg-Strauss o a granulomatosis
broncocéntrica. Un factor reumatoideo positivo obliga a tomar en consideración
la existencia de artritis reumatoidea, así como la positividad de cANCA
apunta a granulomatosis de Wegener.
+ Lavado alveolar: puede resultar
diagnóstico en enfermedades como la granulomatosis de Langerhans.
Sólo cuando no hay ninguna otra
evidencia, consideraremos enfemedades como la sarcoidosis, granulomatosis
necrotizante sarcoidea o la granulomatosis linfomatoide. Se ha descrito
también la presencia de granulomas en asociación con ciertas neoplasias.
En un 20% de los casos no se consigue
establecer la causa de una enfermedad granulomatosa, si bien esto es más
frecuente cuando se encuentran granulomas en biopsias procedentes de hígado
o de ganglios linfáticos. En este caso resulta muy importante el curso
clínico de la enfermedad y también pueden resultar útiles ciertos estudios
de inmunohistoquímica.
APORTACIÓN DEL RADIÓLOGO EN EL DIAGNÓSTICO DE LAS ENFERMEDADES GRANULOMATOSAS PULMONARES
Dra.
Inmaculada Herráez, Servicio de Radiodiagnóstico
¿Cuales
son los hallazgos radiológicos en las granulomatosis pulmonares? ¿Existen patrones radiológicos comunes en estas enfermedades?
Para responder a las dos cuestiones vamos a considerar
dos grandes grupos de enfermedades granulomatosas: infecciosas y no infecciosas.
GRANULOMATOSIS
INFECCIOSAS
Tuberculosis
En la mayoría de los casos en los que se sospecha
la presencia de tuberculosis, la RX de tórax es suficiente para valorar
la afectación pulmonar y la evolución tras el tratamiento (Fig. 1) (1).
Los hallazgos radiológicos más frecuentes en la tuberculosis primaria
son las adenopatías y las condensaciones y en la postprimaria las consolidaciones,
los nódulos y las cavidades, predominantes en los segmentos apicales y
posteriores de los lóbulos superiores (LLSS) (2).
 |
 |
Fig. 1a. Tuberculosis activa. a) RX de tórax: consolidaciones
irregulares en los LLSS (flecha verde) y adenopatías en el hilio izquierdo
(flecha blanca) en un caso de tuberculosis postprimaria. b) RX tórax: patrón nodulillar bilateral en tuberculosis
miliar.
En algunos casos, sin embargo, es muy difícil valorar
si las lesiones radiológicas son activas o no, especialmente si no se
dispone de estudios previos para comparar evolución (Fig. 2). En estos
casos la TC y la TC de alta resolución de tórax (TCAR) pueden ser muy
útiles para valorar la actividad y visualizar lesiones no visibles en
la RX de tórax (Fig. 3) (3). Las lesiones que se encuentran con mayor
frecuencia en TCAR en caso de tuberculosis activa (4) son las que indican
diseminación broncógena: nódulos acinares, lesiones ramificadas y nódulos
centrilobulillares, con distribución típica alrededor y a distancia de
cavidades y consolidaciones pulmonares (Fig. 4). En el caso de tuberculosis
miliar se ven nodulillos pulmonares con distribución al azar (Fig. 5)
(5).
|
Fig. 2. RX de tórax: imágenes lineales irregulares y nodulares en LSD, no siendo posible determinar si se trata de lesiones activas o inactivas. |
 |
 |
 |
Fig. 3. TCAR del paciente de la Fig. 2. Lesión cavitada
(flecha verde) con nódulos y lesiones ramificadas satélites (punta de
flecha) en segmento posterior de LSD, nódulos acinares en segmento anterior
de LSD (flecha blanca) y zonas parcheadas de opacidad en vidrio deslustrado
en LSI (flecha negra), todo ello indicativo de actividad.
 |
 |
Fig. 4. Tuberculosis postprimaria. TCAR de otro paciente
con lesiones características de diseminación broncógena: cavidades (flecha
negra) y consolidaciones (flecha verde) en LSI; nódulos acinares y centrilobulillares
(puntas de flecha) y lesiones ramificadas (flecha blanca) alrededor y
a distancia de las cavidades y consolidaciones, con afectación multilobar.
|
Fig. 5. Tuberculosis miliar. TCAR: nodulillos (puntas de flechas) con distribución al azar y mayor afectación de LLSS. Las lesiones no eran visibles en RX de tórax. |
 |
Mycobacterias
no tuberculosas
Los hallazgos radiológicos son similares a los de
la tuberculosis, aunque son más frecuentes las bronquiectasias, afectando
típicamente el LMD y la língula ( Fig. 6) (6).
|
Fig. 6. Infección por Mycobacterium gordonae. TCAR: en LLSS se ven nódulos, uno de ellos cavitado y otro rodeado de opacidad en vidrio deslustrado (signo del halo) (flecha negra). Se ven también zonas parcheadas de vidrio deslustrado (punta de flecha) y bronquiectasias (flecha blanca). |
 |
Neumonía
por Pneumocystis carinii
La
afectación radiológica típica en esta infección oportunista de pacientes
inmunodeprimidos es la consolidación perihiliar, bilateral y simétrica,
que posteriormente se extiende al resto del parénquima (Fig. 7) (7).
|
Fig. 7. Neumonía por P. Carinii en paciente VIH +. RX de tórax: tenues condensaciones perihiliares y patrón reticular en la periferia. |
 |
Habitualmente
la RX de tórax es suficiente para sugerir el diagnóstico. No obstante,
si hay sospecha clínica de la enfermedad y la RX de tórax es normal o
indeterminada, la TCAR es muy útil, con una sensibilidad del 100% y una
especificidad del 89%. Las lesiones encontradas con mayor frecuencia son
áreas bilaterales de opacidad en vidrio deslustrado, difusas o en mosaico
(8). Se pueden ver también consolidaciones, quistes, nódulos y septos
interlobulillares engrosados (Fig. 8).
|
Fig. 8. Neumonía por P. Carinii en paciente VIH +. TCAR: afectación bilateral con zonas parcheadas y nodulares de opacidad en vidrio deslustrado (puntas de flecha) y pequeños quistes (flecha). |
 |
GRANULOMATOSIS NO INFECCIOSAS
Es una rara enfermedad multisistémica que ocurre casi exclusivamente
en asmáticos. La RX de tórax está alterada en el 70% de los pacientes.
Sin embargo, los hallazgos radiológicos no son específicos, ni en RX de
tórax ni en TCAR, siendo necesario interpretarlos en el contexto clínico
adecuado (asma, eosinofilia, rinosinusitis alérgica, neuropatía). Los
hallazgos más frecuentes son las consolidaciones y las opacidades en vidrio
deslustrado parcheadas, periféricas, no segmentarias, transitorias (9).
Con menor frecuencia se pueden ver nódulos centrilobulillares, que no
suelen cavitar (Fig. 9).
 |
 |
||
Fig. 9. S. de
Churg-Strauss. TCAR: nódulos
centrilobulillares y peribronquiales (flechas) y pequeñas zonas de opacidad
en vidrio deslustrado en pulmón derecho .
Enfermedad de Wegener
Es otra rara enfermedad multisistémica en la que
existe afectación granulomatosa de los tractos respiratorios superior
e inferior, glomerulonefritis y vasculitis necrotizante de múltiples órganos
y tejidos. Hay una forma limitada al tracto respiratorio. Se detectan
anticuerpos C-ANCA en el 90% de los casos.
En este contexto clínico, la RX de tórax inicial
muestra alteraciones en el 45% de los casos y los hallazgos más frecuentes
son los nódulos bilaterales que, al contrario que en el síndrome de Churg-Strauss,
cavitan en el 50% de los casos (Fig. 10) (9).
|
Fig. 10. Enfermedad de Wegener. RX de tórax: nódulos en ambos campos pulmonares. |
 |
La
TCAR es útil para demostrar lesiones no visibles en RX de tórax y para
sugerir la existencia de actividad de la enfermedad; en el 60% de los
pacientes con enfermedad activa se ven nódulos, masas o condensaciones,
mientras que estas alteraciones sólo se ven en el 20% de los casos inactivos
(Fig. 11) (10).
 |
 |
||
Fig.
11. Enfermedad de Wegener. a)
TC: masa cavitada en segmento
apical de LII. b) TCAR: nódulos y condensaciones lobulillares con broncograma
aéreo (flecha blanca), bilaterales.
Artritis
reumatoide
La afectación granulomatosa pulmonar en la artritis
reumatoide ocurre en forma de nódulos necrobióticos o reumatoides, similares
a los subcutáneos (11). En RX de tórax se ven en el 0,2% de los pacientes
y además del pulmón pueden afectar la pleura y el pericardio (Fig. 12).
|
Fig. 12. Artritis
reumatoide. RX de tórax:
nódulos y masas lobuladas periféricas, con predominio en hemitórax derecho. |
 |
En
 |
 |
Fig. 13. Nódulos necrobióticos en artritis reumatoide.
TCAR: nódulos periféricos y subpleurales bilaterales (flecha negra), algunos
cavitados (flecha verde).
Sarcoidosis
La
mayoría de la morbi-mortalidad
de la enfermedad se debe a la afectación pulmonar.
Radiológicamente
se distinguen cinco estadios, con importancia pronóstica y terapéutica:
0) sin afectación; 1) con afectación ganglionar; 2) con afectación ganglionar
y parenquimatosa pulmonar; 3) con afectación parenquimatosa pulmonar y
4) fibrosis pulmonar.
Los granulomas sarcoideos se distribuyen a lo largo
de los linfáticos en los haces broncovasculares y, en menor medida, en
los septos interlobulillares y regiones subpleurales, siendo ésta la causa
del alto rendimiento diagnóstico de la biopsia transbronquial. La distribución
de las lesiones parenquimatosas es más difícil de apreciar en RX de tórax,
pero es clara en TCAR (Fig. 14 y 15), donde se observa engrosamiento intersticial
peribroncovascular de contorno liso o nodular, de predominio en las zonas
perihiliares, como hallazgo más frecuente. Se pueden ver también nódulos
subpleurales, centrilobulillares y en septos interlobulillares (13).
 |
 |
Fig. 14. Sarcoidosis. Las radiografías de tórax PA y L muestran
un patrón reticulonodular perihiliar bilateral.

Fig. 15. Sarcoidosis.
TCAR del mismo paciente de la
Fig. 14. Engrosamiento del intersticio peribroncovascular central, bilateral,
con contorno nodular (flecha verde). Se ven también nódulos centrilobulillares
(flecha negra), subpleurales y en septos interlobulillares (flecha blanca).
En cortes de TCAR realizados en espiración es frecuente
la aparición de zonas parcheadas de atrapamiento aéreo, que reflejan la
afectación de la pequeña vía aérea (Fig. 16).
 |
 |
Fig. 16. Sarcoidosis. a) TCAR en inspiración: nodulillos
centrilobulillares (flecha blanca) y subpleurales (flechas verdes) con
distribución parcheada; en algunos grupos se asocia opacidad en vidrio
deslustrado. b) TCAR en espiración: en un corte al mismo nivel que en
inspiración se ven zonas parcheadas de menor atenuación por atrapamiento
aéreo (flecha negra).
El patrón y la extensión de las lesiones vistas en
TCAR se correlacionan de forma imprecisa con las pruebas de función respiratoria
(PFR), por lo que en el seguimiento de los pacientes con sarcoidosis la
TCAR estaría indicada solamente en los casos con RX de tórax normal y
PFR alteradas, pacientes con hemoptisis, con sospecha de una segunda enfermedad
y en los candidatos a transplante.
La sarcoidosis puede evolucionar hacia la fibrosis
(estadio 4) (Fig. 17).
|
Fig.
17. Fibrosis pulmonar en un caso de
sarcoidosis. La TCAR muestra patrón
reticular y pequeños quistes de
panal subpleurales (flecha negra), distorsión
del parénquima pulmonar y
bronquiectasias de tracción (flecha
blanca). |
 |
Alveolitis
alérgica extrínseca
Los
hallazgos radiológicos son distintos en los diferentes estadios de la
enfermedad e independientes del tipo de alérgeno inhalado (14).
En
la fase aguda predomina la consolidación.
En
la fase subaguda, la RX de tórax puede mostrar un patrón nodular predominante
en campos medios e inferiores o puede ser normal. En TCAR se ven áreas
bilaterales de opacidad en vidrio deslustrado y nódulos centrilobulillares
mal definidos, con predominio en las mismas zonas; en los cortes realizados
en espiración se pueden ver áreas de atrapamiento aéreo (Fig. 18 y 19).
En estas fases los hallazgos son reversibles y, en el contexto clínico
adecuado, muy sugestivos de la enfermedad.
En
la fase crónica se observan signos de fibrosis, tanto en RX de tórax como
en TCAR.
 |
 |
Fig.
18. Alveolitis alérgica extrínseca en fase subaguda. a) La TCAR en inspiración
muestra zonas parcheadas de opacidad en vidrio deslustrado con el signo
del bronquio negro. b) En espiración se observan zonas también parcheadas
de atrapamiento aéreo. La RX de tórax era normal.
 |
 |
Fig. 19. Alveolitis alérgica extrínseca subaguda. Presentación inusual en RX de tórax y TCAR, con nódulos con signo del halo debidos a extensa reacción tipo bronquiolitis obliterante con neumonía organizada. La afectación es mayor en campos medios e inferiores.
CONCLUSIONES
En la sarcoidosis
los hallazgos principales son los nódulos con distribución perilinfática,
con mayor frecuencia peribroncovasculares centrales.
Los hallazgos
de TCAR pueden sugerir el diagnóstico en el contexto clínico adecuado.
BIBLIOGRAFÍA
1. Bass JR Jr, Farer
LS, Hopewell PC, Jacobo RF, Snider DE Jr. Diagnostic standars and classification
of tuberculosis. Am Rev Respir Dis 1990; 142: 725-735.
2. Müller NL, Fraser
RS, Colman NC, Paré PD. Pulmonary infection. En: Radiologic diagnosis
of diseases of the chest. 1ª ed. Philadelphia. Saunders. 2001, pag. 156-167.
3. Leung AN. Pulmonary
tuberculosis. The essentials. Radiology 1999; 210:307-322.
4. Im J-G, Itoh H,
Shim Y-S, Lee JH, Ahn JA, Han MCh et al. Pulmonary tuberculosis: CT findings-
early active disease and sequential change with antituberculous therapy:
Radiology 1993; 186: 653-660.
5. Oh Y-W, Kim YH,
Lee NJ, Kim JH, Chung KB, Suh WH et al. High-resolution CT appearance
of miliary tuberculosis. J Comput Assist Tomog 1994; 18 (6): 862-866.
6. Miller Jr WT.
Spectrum of pulmonary nontuberculous mycobacterial infection. Radiology
1994; 191: 343-350.
7. Boiselle PM, Crans
Jr CA, Kaplan MA. The changing face of Pneumocystis carinii pneumonia
in AIDS patients: AJR 1999; 172: 1301-1309.
8. Webb WR, Müller
NL, Naidich DP. Diseases characterized primarily by parenchymal opacification.
En: High-resolution CT of the lung. 3ª ed. Philadelphia. Lippincott Williams
& Wilkins. 2001, pag. 396- 403.
9. Mayberry JP, Primack
SL, Müller NL. Thoracic manifestations of systemic autoinmune diseases:
radiographic and high-resolution CT findings. Radiographics 2000; 20:1623-1635.
10. Webb WR, Müller
NL, Naidich DP. Diseases characterized primarily by nodular or reticulonodular
opacities. En: High-resolution CT of the lung. 3ª ed. Philadelphia. Lippincott
Williams & Wilkins. 2001, pag. 314-315.
11. Katzenstein AL
A. Systemic diseases involving the lung. En: Katzenstein and Askin´s surgical
pathology of non-neoplastic lung disease. 3ª ed. Philadelphia. W. B. Saunders.
1997, pag. 168-170.
12. Carotti M, Salaffi
F, ManganelliP, Salvolini L, Bichi Secchi E, de Berardinis S. The subclinical
involvement of the lung in rheumatoid arthritis: evaluation by high-resolution
CT. Reumatismo 2001; 53 (4): 280-288.
13. Halme M, Piilonen
A, Taskinen E. Comparison of endobronchial and transbronchial biopsies
with high-resolution CT in the diagnosis of sarcoidosis. APMIS 2001; 109
(4): 289-294.
14. Patel RA, Sellami
D, Gotway MB, Golden JA, Webb R. Hypersensitivity pneumonitis: patterns
on high-resolution CT. J Comp Assist Tomog 2000; 24 (6): 965-970.
PATOGÉNESIS
Y PARAMETROS HISTOLÓGICOS EN EL DIAGNOSTICO DIFERENCIAL
Dra.
Nieves Alonso, Servicio de Anatomía Patológica
PATOGÉNESIS DE LA
INFLAMACIÓN GRANULOMATOSA
Organismos
vivos, partículas orgánicas o inorgánicas, insolubles o lentamente eliminables,
inducen la formación de granulomas. En el pulmón, dichos agentes llegan
por vía inhalatoria o circulatoria.
La respuesta inflamatoria
granulomatosa, es regulada por mecanismos inmunológicos o por mecanismos
filogenéticos primarios de fagocitosis y degradación lisosomal.
La formación de granulomas
por mecanismos inmunes puede progresar por un perfil de linfocitos T-helper-1,
linfocitos T-helper-2 y linfocitos T citotóxicos.
Las células TH1 producen factor de necrosis( TNFa) Interleuquina (IL3), (IL12) e interferón g (IFNg)
activador de macrófagos,promoviendo la inmunidad celular.
Las células TH2 expresan interleuquinas (IL4), (IL5), e (IL10), que son importantes en la respuesta mediada por anticuerpos y procesos alérgicos.
Las
células TH1 y TH2, muestran una regulación cruzada. El IFNg, disminuye la producción de citoquinas y la proliferación
de TH2, y la IL4 IL10, induce descenso en la producción de IFNg por las células TH1.
CLASIFICACION
DE LA INFLAMACION GRANULOMATOSA
Intentos
de clasificación aúnan aspectos morfológicos y funcionales para el mejor
entendimiento del proceso.
- Según patrón
morfológico se distinguen granulomas epiteliodes y de cuerpo extraño.
- Según cinética
celular:
Granulomas con bajo recambio celular producidos por agentes inertes pobremente degradables y no tóxicos para la célula. Los macrófagos acumulan gran cantidad del agente irritante y en ocasiones se constituyen granuloma de cuerpo extraño.
Granulomas con alto índice de recambio celular, son
producidos por agentes tóxicos para la célula, tales como las micobacterias.
Se caracterizan por un alto índice de división local para compensar la
muerte celular y el agente causal
sólo se encuentra en una pequeña proporción de células. En la practica
clínica la mayoría pertenecen a este grupo y los macrófagos muestran una
evidente heterogeneidad funcional.
- Según participación
de respuesta inmunológica influyendo el estado inmunológico del
huésped en determinar la morfología del granuloma.
HISTOLOGIA
DE LA INFLAMACION GRANULOMATOSA
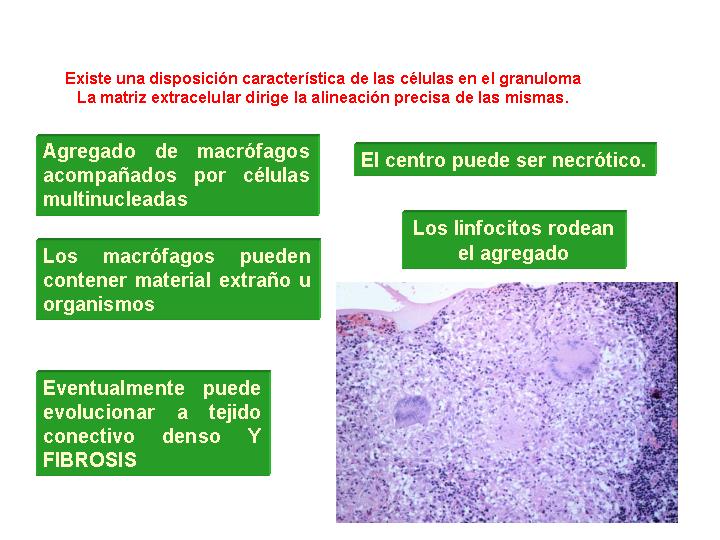
El granuloma está
definido como una lesión inflamatoria nodular bien circunscrita, compuesta
por diferentes células, que según el tipo predominante se pueden sistematizar
en:
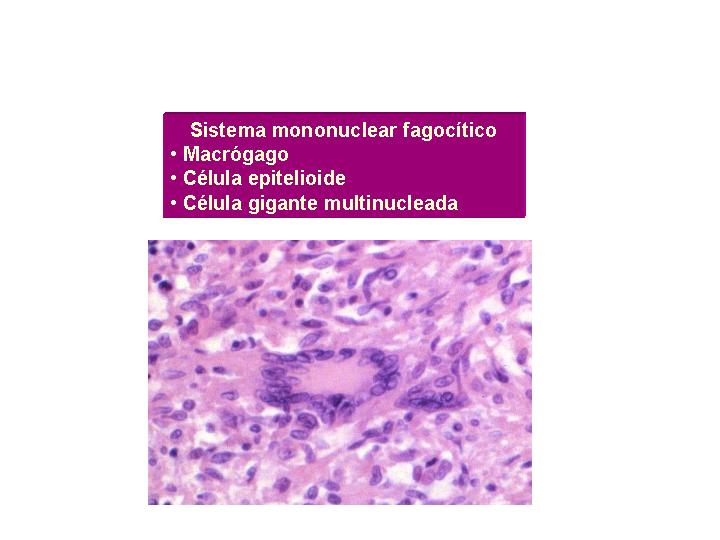
1.- Histiocitos Granulomatosis
histiocítica.
2.- Células epiteliodes
y células gigantes Granuloma epiteliode o sarcoideo.
3.- Células de Langerhans
Granulomatosis de células de Langerhans.
4.- Células gigantes
de cuerpo extraño Granuloma de cuerpo extraño.
5.- Fibroblastos con
hialinización Granuloma hialinizante.
A su vez puede asociar una mezcla de eosinófilos, neutrófilos,
linfocitos y plasmáticas.(Fig1
y 2 )
{kind=link}
{kind=link}
Los granulomas pueden
presentar necrosis central por diferentes mecanismos:
1.- Trombosis o vasculitis
seguido de necrosis isquémica.
2.- Mediadores químicos
que inducen a agregación plaquetaria y necrosis coagulativa.
3.- Mecanismos de
apoptosis o por perforinas liberadas por células natural killer.
DIAGNOSTICO DIFERENCIAL
Sistematizamos el diagnóstico diferencial confrontando las
diferentes etiologías con el patrón morfológico del granuloma.( Fig. 3 )
{kind=link}
.
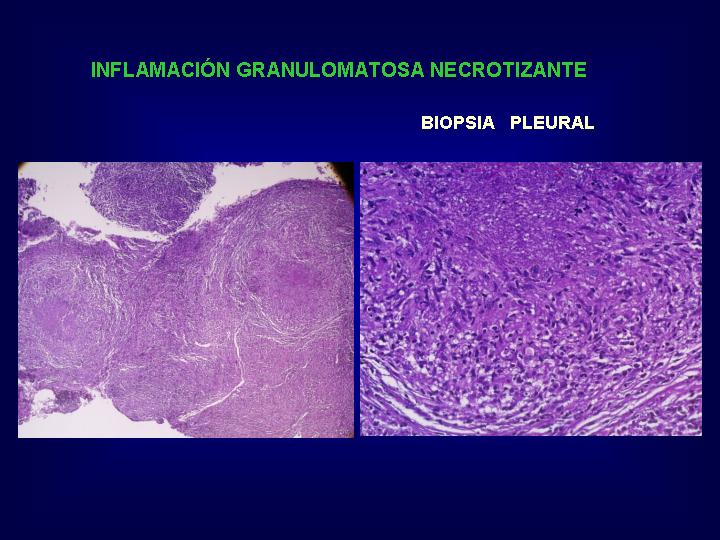
1.- GRANULOMAS EPITELIODES
O SARCOIDEOS
La imagen más específica
de este tipo de granuloma, lo constituye la interacción entre linfocitos
y célula epiteliode.(
Fig.4 )
{kind=link}
Tres tipos de agentes
conocidos causan granuloma epitelioides:
- Organismos infecciosos:
- Hongo:
Pneumocistis
carinii
Aspergilosis,
blastomicosis.coccidiomicosis, criptococosis,histoplasmosis
- Bacterias:
mycoplasma
Bacilos
ácido-alcohol resistentes (micobacterium tuberculosis, m. atípicas)
- Productos de plantas y animales: (polen, proteinas,
esporangios)
- Compuestos metálicos: ( aluminio, berilio, bario,cobalto,
cobre, oro,lantánidos, titanio y zirconio)
: a). GRANULOMAS INFECCIOSOS:
Los organismos infecciosos,
producen más de la mitad de granulomas vistos en la práctica de la patología
quirúrgica.
En el protocolo de
estudio deben incluirse técnicas especiales y/o de cultivo para definir
el agente causal.
·
Tinciones ácido-alcohol
resistentes y estudios de PCR ( reacción en cadena de polimerasa ), para
detectar micobacterias en granulomas no necrotizantes o hialinizados.
·
Tinción de plata e inmunohistoquímica
para el estudio de hongos.
·
Técnicas específicas
para infecciones parasitarias.
Los
granulomas infecciosos pueden
ser solitarios o múltiples, broncocéntricos, alveolares o linfangíticos
en su distribución, y necrotizantes o no necrotizantes, o incluso mixtos.
La estrategia diagnóstica
persigue demostrar el microorganismo en el tejido.
Los hongos son más
fácilmente vistos en cortes histológicos que los bacilos ácido alcohol
resistentes. Algunas infecciones tales como Coccidiomicosis y Blastomicosis, producen granulomas necrotizantes
más supurativos en los cuales el infiltrado neutrofílico acompaña a los
elementos necróticos. En ocasiones H. Capsulatum a diferencia de muchos
otros hongos, pueden no ser visibles dentro de los granulomas con las
técnicas convencionales de hematoxilina-eosina , y requieren prolongadas
tinciones con plata-metenamina.
Raramente infecciones
con pneumocystis carinii, puede producir una respuesta inflamatoria granulomatosa
necrotizante. Esta manifestación ha sido vista preferentemente en pacientes
inmunosuprimidos. Microscópicamente la mayoría de los granulomas muestran
necrosis irregular con un material
eosinofílico reminiscente del exudado intraalveolar visto en la típica
neumonía por P. Carinii, y en estos casos una cuidadosa búsqueda de microorganismos,
usando técnicas de plata o técnicas inmunohistoquímicas, pueden ser necesarias
para el diagnóstico.
Infecciones por micobacterias
tienen una especial relevancia
epidemiológica, dado que estudios recientes de la OMS consideran que un
tercio de la población mundial, está infectada, si bien solo un 10 % desarrollan
enfermedad clínica según susceptibilidades genéticas.
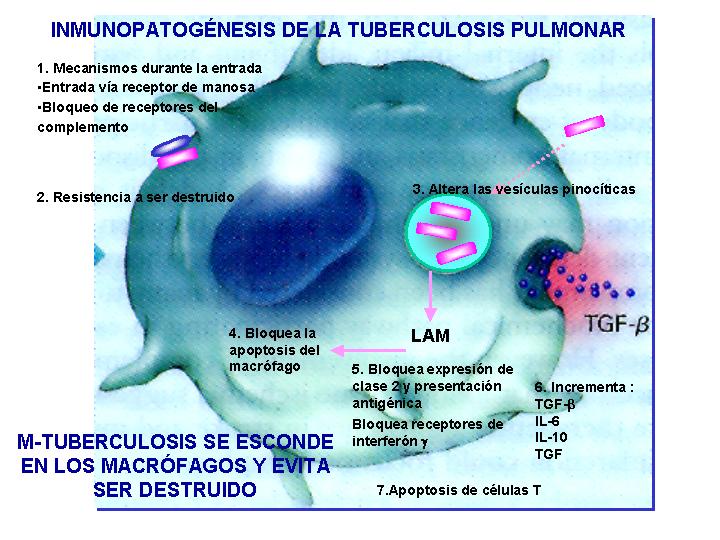
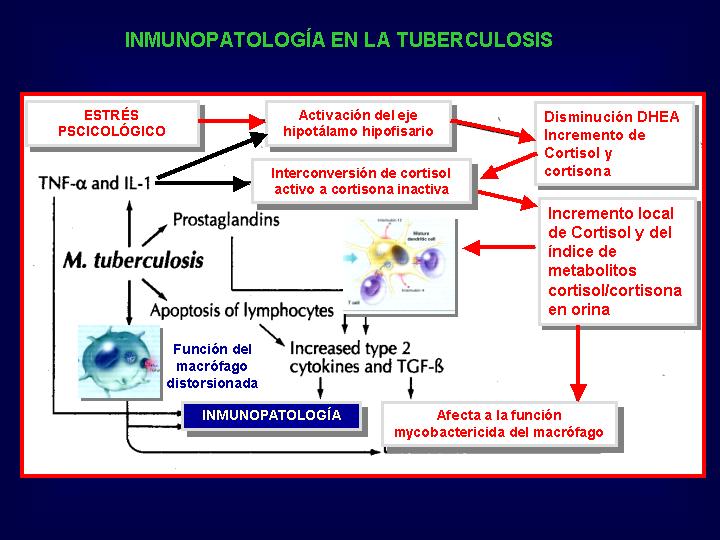
Inmunopatogénesis de la tuberculosis pulmonar
Las micobacterias patógenas sintetizan una molécula C4 like
que lleva al depósito de C3B sobre la membrana celular de la micobacteria,
e incrementa el paso de las mismas dentro del macrófago.Una vez dentro
del macrófago su resistencia frente a las defensas del organismo es debido
a su capacidad para inhibir la fusión de fagolisosomas y sobrevivir en
los fagocitos del huésped, inhibiendo a su vez su apoptosis.(Fig.5
)
{kind=link}
La virulencia de la
micobacteria se correlaciona con su capacidad de crecimiento intracelular
e inducción de TNFa. Si dicha respuesta es sostenida puede ser perjudicial
y llevar a la consolidación y daño tisular.
Una respuesta inmunológica
útil frente a la micobacteria requiere un balance entre : respuesta potente
antimicrobiana, desarrollo de la memoria inmunológica y control de respuesta
inflamatoria.(Fig.6
y 7 )
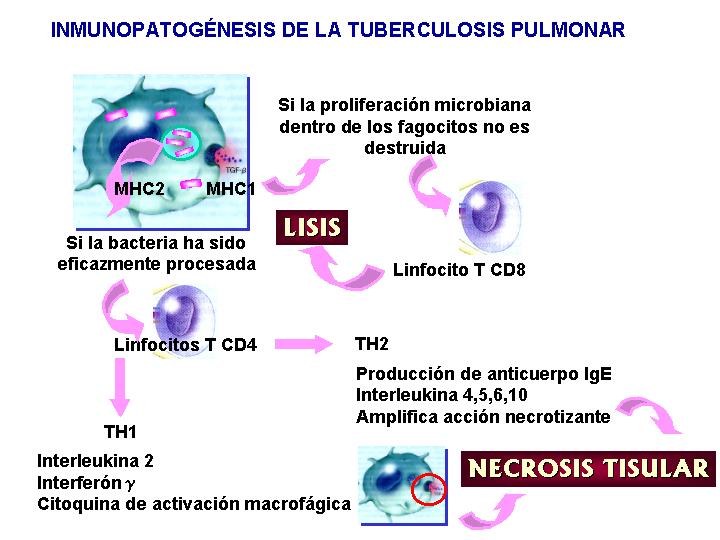{kind=link}
{kind=link}
En pacientes inmunocompetentes,
infecciones por micobacteria no tuberculosa (m. avium) puede causar una
neumonitis granulomatosa no necrotizante sarcoidea. Son especialmente
susceptibles pacientes con enfermedad crónica de vía aérea; las bronquiectasias
son colonizadas por micobacterium avium desarrollándose una infección
de bajo grado que se manifiesta como enfermedad pulmonar difusa con granulomas
sarcoideos que pueden mostrar bronquiolocentricidad o estar distribuidos
al azar, sin seguir un patrón linfangítico.Los granulomas típicamente
tienen una corona de linfocitos y el parénquima pulmonar intercalado es
relativamente normal.(
Fig. 8 )
{kind=link}
b).
GRANULOMATOSIS INDUCIDAS POR METALES
Diferentes metales
poseen propiedades antigénicas o físico-químicas que promueven el desarrollo
de granulomas sarcoideos en el pulmón.
Fisiopatología:
- Inmunidad mediada por células antígeno-específico
- Demostrada: *berilio,titanio,zirconio.
- Posible: aluminio,cobalto y oro.
- Reacción de tipo cuerpo extraño: aspiración
de bario
- Desconocido: lantánidos.
*La respuesta inmunológica
al berilio es considerada como modelo de enfermedad granulomatosa mediada
inmunológicamente.Clones de células T específicas aparecen precozmente
en el curso de la enfermedad, produciendo un patrón de citoquinas tipo
TH-1 Varios estudios, sugieren que (HLA)-DP (human lymphocyte antigen),
está implicado en la aparición de clones de células T reactivas al berilio
. Una sustitución alélica del ácido glutámico en posición 69 del gen HLA-DP,
se asocia a un riesgo mayor para desarrollar sensibilización al berilio.
Metodología de estudio:
La mayoría de las
partículas de metal pueden ser identificadas en el tejido, utilizando técnicas de difracción para microanálisis
de rayos X y microscopia electrónica
de barrido .
Se complementa con
test inmunológicos de sensibilización.
En general las lesiones
están constituidas por agregados de células inmunoefectoras con un anillo
periférico en el granuloma maduro que contiene gran número de mastocitos,
implicados en la proliferación
fibroblástica. Se observan patrones histológicos análogos a los de la
sarcoidosis. (Fig. 9 )
{kind=link}
C).-ALVEOLITIS
ALERGICA EXTRINSECA
Resulta de la inhalación de una amplia variedad de sustancias orgánicas y químicas que son capaces de actuar como antígenos extraños e inducir una reacción de hipersensibilidad.
Dependiendo
de la cantidad de antigeno inhalado, frecuencia de exposición y factores
del huesped, varía la forma de presentación clínica. Pacientes con una
exposición baja pero permanente desarrollan un cuadro insidioso de insuficiencia
respiratoria y son los candidatos a una posible biopsia pulmonar. Test
cutaneos y serológicos ayudan a confirmar el diagnóstico, pero dado que
hay más antígenos en la naturaleza que pruebas inmunológicas, este puede
no ser identificado.
Patogénesis:
Se
ha considerado que están implicados
dos mecanismos patológicos:
a.-
Respuesta mediada por inmunocomplejos que inducen un daño pulmonar agudo
via complemento, dependiente de neutrófilos.
b.-
Respuesta mediada por células T que inducen inflamación granulomatosa.(Fig.10 )
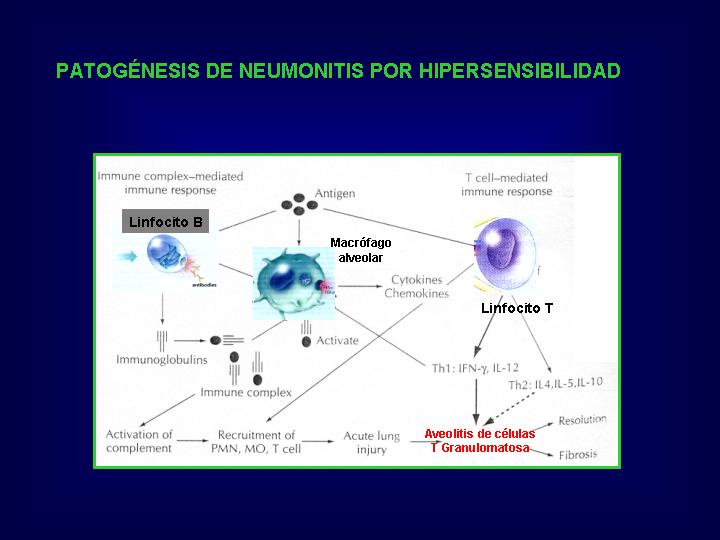{kind=link}
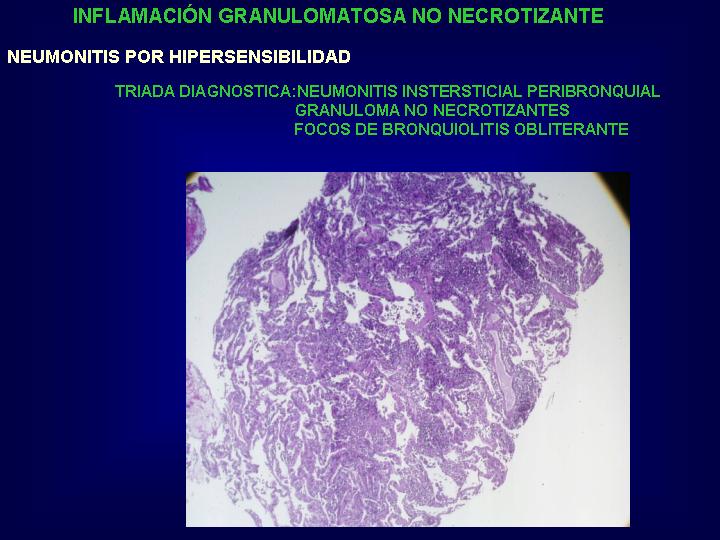
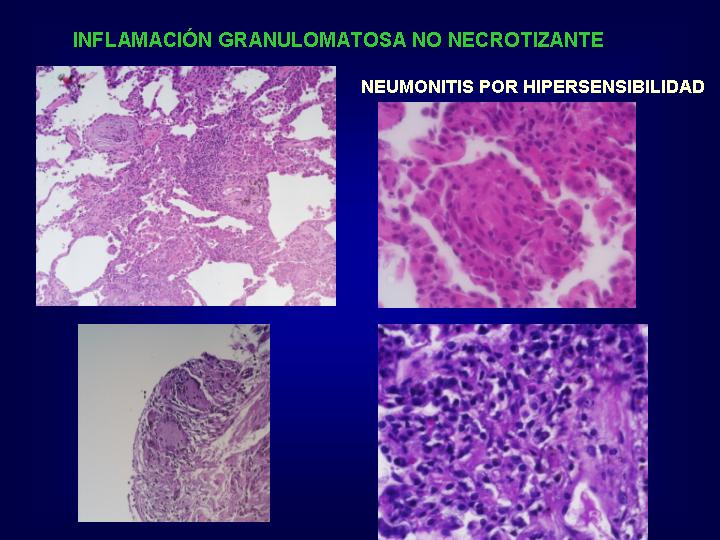
Se establece una neumonitis
intersticial granulomatosa con un grado variable de bronquiolitis
obliterante y expansión en el pulmón de células T CD8 citotóxicas.
Alteraciones específicas
en el receptor antigénico de células T (TCR) y defectos en la capacidad
de los macrófagos para modificar la actividad linfoproliferativas se consideran
responsables de la alveolitis linfocitaria observada en la neumonitis
de hipersensibilidad.
El proceso inflamatorio
tiende a mostrar una distribución peribronquiolar diferenciandolo de la
neumonía intersticial linfoide que muestra una infiltración linfocitaria
más intensa y más difusa con granulomas pobremente formados.
Los granulomas son
más pequeños y menos frecuentes que en la sarcoidosis. Las adenopatías
hiliares no están afectadas y las células gigantes poseen hendiduras aciculares
que sugieren el diagnóstico sin total especificidad.
La organización de
exudados luminares dentro de alveolos y bronquiolos son los responsables
de la acumulación de lípidos en los macrófagos produciendo un patrón de
neumonía lipoidea endógena.(Fig.
11 y 12 )
{kind=link}
{kind=link}
D).-SARCOIDOSIS
Enfermedad multisistémica
granulomatosa, constituye una entidad de diagnóstico clínico patológico
Después de la exclusión
de otras etiologías a través de la historia clínica (ambiental, ocupacional,
médica), estudios microbiológicos, radiografía y estudios serológicos
se establece el diagnóstico.
Patogénesis:
Técnicas de biología
molecular y estudios inmunológicos, han despertado una antigua controversia
que asocia sarcoidosis con agentes infecciosos tipo micobacterias y organismos
propionibacterias. Dada la similitud observada en el perfil de citoquinas
inducida por dichos agentes y el observado en la sarcoidosis.
Aspectos epidemiológicos
han establecido una relación positiva con la exposición a insecticidas
,pesticidas, tierra vegetal y moho, los cuales son también marcadores
de ambientes ricos en agentes microbianos.
El hallazgo más relevante
en la etiopatogénesis, lo constituye la asociación con una respuesta inmunológica
TH1 intensificada, ligada a un número limitado de patógenos y asociado
a una predisposición genética.Concordante con esta respuesta TH1 dominante,
se ha observado una elevación en IFNg (factor activador de macrófagos).
La formación del granuloma
sarcoideo es el resultado final de la interrelación compleja de muchos
factores, incluyendo :antígeno invasor, antigenémia persistente, presentación
macrofágica, reconocimiento de células TH1, hiperactividad de célula B,
aparición de inmunocomplejos circulantes y citoquinas específicas.
De esta forma se llega
al estado de aberración inmunológica que subyace en la sarcoidosis:
- Inmunidad celular periférica deprimida.
- Respuesta granulomatosa TH1 hiperactiva.
- Respuesta humoral intensificada.
En general esta enfermedad
afecta a individuos con sistemas inmunológicos competentes que son capaces
de protegerse del agente patógeno, pero son incapaces de regular una respuesta
inmunológica exuberante debido al fallo de mecanismos inhibidores como
la apoptosis.
En el campo de las
enfermedades intersticiales pulmonares, la sarcoidosis es considerada
como paradigma de enfermedad inmunomediada y parámetros inmunológicos
son claves en la evolución y en la extensión de la enfermedad. En el análisis
de citometría de flujo de lavado broncoalveolar, el perfil fenotípico
de células T, sin llegar a tener especificidad diagnóstica, si ha mostrado
significado pronóstico; hay estudios que indican como parámetro favorable
un incremento en el BAL del indice CD4/CD8 en particular si está asociado
con fenotipo HLA DR 17.
El patrón histológico
muestra granulomas sarcoideos multifocales de apariencia uniforme en distribución
linfangítico en relación con estructuras bronquiolo arteriales, septos
interlobulares y en la periferia del pulmón subpleurales.(Fig.13 )
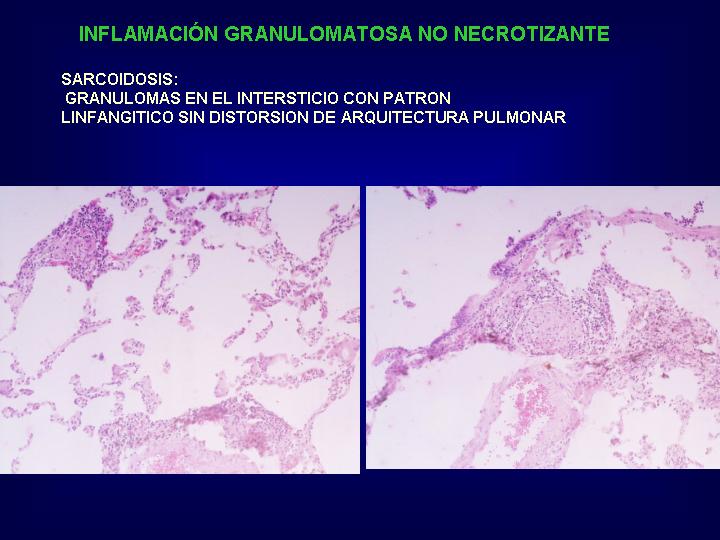{kind=link}
Es frecuente la
afectación de vía aérea con un patrón de bronquiolitis granulomatosa,
y aunque la inflamación pueda afectar a pequeña vía aérea, imágenes de
bronquiolitis obliterante son infrecuentes, siendo más típicas de neumonitis
de hipersensibilidad , reacción a drogas o infecciones .
Los granulomas sarcoideos
tienen una pronunciada distribución vasculocéntrica, siendo las
venas las más frecuentemente afectadas, dañando preferentemente la adventicia
y parte externa de la media, lo cual plantea diagnóstico diferencial con
otras vasculitis granulomatosas.
Otro aspecto diagnóstico
lo constituyen estructuras citoplasmáticas inespecíficas(Fig.14) identificadas
dentro del granuloma. Cuerpos de Shaumann, cuerpos asteroides de estructura
espiculada o cristales de oxalato cálcico birrefringente, que pueden inducir
a un error de interpretación como granulomas de cuerpo extraño.
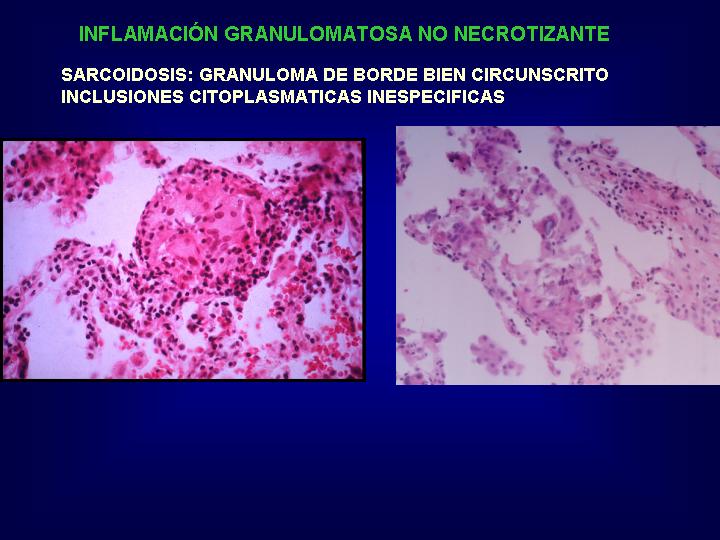{kind=link}
Variantes histológicas:
Sarcoidosis nodular: los granulomas pueden ser tan
numerosos que se hacen confluyentes y se constituyen grandes masas de
tejido sarcoideo que sugieren enfermedad metastásica en la radiología.
En general son pacientes jóvenes con adenopatía hiliares bilaterales y
un pronóstico favorable hacia la resolución espontánea.
Granulomatosis sarcoidea necrotizante: asocia neumonitis
granulomatosa con patrón linfangítico, necrosis variable y vasculitis
en arterias pulmonares y venas, con un patrón de granuloma necrotizante,
arteritis de células gigantes o infiltrados inespecíficos de linfocítos
y macrófagos.
Esta variante puede
mostrar bronquiolitis obliterante granulomatosa y en algunos casos está
asociada a otros trastornos inmunológicos como enfermedad inflamatoria
intestinal o síndrome de Sjögren.
Patrón de reacción
inespecífica en:
Asociación con procesos malignos: los granulomas sarcoideos
pueden ser vistos adyacentes a un carcinoma primario de pulmón y enmascarar
el diagnóstico del tumor.Forma parte de reacciones sarcoideas relacionadas
con procesos tumorales, bien por carcinomas, seminoma,disgerminoma o procesos
linfoproliferativos.
Asociación
con procesos linfoproliferativos pulmonares, preferentemente linfomas
B de bajo grado.Linfocitos pequeños y maduros proliferan alrededor de
haces broncovasculares, engruesan septos interlobulares y finalmente invaden
pared alveolar llegando a obliterar el espacio aéreo; presentando imágenes
de esclerosis hialina, folículos linfoides prominentes, lesiones linfoepiteliales
y ocasionales granulomas de células epiteliodes.
Asociación
a enfermedades del colágeno, más frecuentemente en el Síndrome de Sjögren,
en el contexto habitual de una neumonía intersticial linfoide.
Finalmente
en lesiones granulomatosas de significado desconocido.(Sindrome GLUS)
Inmunohistoquímicamente, los granulomas están constituidos por células
B positivas como en las reacciones sarcoideas a tumores, sin embargo en
la sarcoidosis son células T.
VASCULITIS NO INFECCIOSAS Y GRANULOMATOSAS
GRANULOMATOSIS DE WEGENER
Vasculitis sistémica primaria pauci-inmune de pequeño vaso
asociada a anticuerpos anticitoplasma neutrofílico.
La presentación
clínica incluye formas clásicas,
limitadas y atípicas. En la forma clásica hay una fase inicial de inflamación
granulomatosa del tracto respiratorio, seguida de una generalización sistémica
de curso fulminante, en un tiempo variable, de semanas a años. En su forma
más típica constituye un síndrome pulmonar renal que va de una microhematuria
asintomática hacia una glomerulonefritis rápidamente progresiva con fallo
renal e infiltrados pulmonares asintomáticos o nódulos que tienden a cavitarse
y a convertirse en focos de infeccion secundaria con neumonías bacterianas.
También puede comenzar como una forma pura de vasculitis de pequeño vaso
en ausencia de lesión granulomatosa, constituyendo un síndrome hemorrágico alveolar.
Formas limitadas con
afectación del tracto respiratorio sin afectación renal
Variantes de la enfermedad:
Formas limitadas con predominante inflamación granulomatosa del tracto
respiratorio superior en ausencia de enfermedad renal. También hay formas
limitadas de inflamación granulomatosa y/o vasculitis afectando a órganos
aislados, tales como órbita o tracto gastrointestinal. Representan formas
frustradas de WG.
WG-ANCA positivo:
menos del 5% de los pacientes son MPO-ANCA positivos (antimieloperoxidasa),
mientras que la gran mayoría es PR3-ANCA positivo.El primer grupo tiene
una afectación más leve y en el segundo
grupo las manifestaciones renales y de órganos extrarenales son observadas
más frecuentemente . Ambas positividades juegan un papel en la fisiopatología
del Wegener clásico y de la poliangitis microscópica.
WG-ANCA negativo: un pequeño grupo de pacientes con Wegener generalizado
permanecen como ANCA negativo. Este grupo presentan prominentes manifestaciones
del tracto respiratorio, parálisis de nervios craneales e inflamación
granulomatosa de las meninges. Esta variante incorpora solo ciertos mecanismos patogénicos que están actuando
en el WG clásico.
Sindrome pseudo-Wegener:
el síndrome incluye extensa destruccción de cartílago nasal y hueso, vasculitis
de pequeño vaso y infecciones respiratorias recurrentes con desarrollo
de bronquiectásias. El deterioro clínico aparece con la instauración de
terapia inmunosupresora. Hay ausencia de PR3-ANCA y MPO-ANCA. Las lesiones
granulomatosas muestran una pronunciada infiltración de células natural
killer y la patogénesis de la enfermedad es claramente diferente de WG,
pese a compartir aspectos clínicos. En esta enfermedad hay una baja expresión
de moléculas HLA I, que favorece la expansión de poblaciones de células
T natural killer autoreactivas en sangre periférica y en las lesiones
granulomatosas observadas.
Asociación de WG
con enfermedad granulomatosa intestinal
PATOGENESIS:
Ademas del valor diagnóstico, los anticuerpos antineutrofílicos
citoplasmáticos tienen un papel importante en la inmunopatogenesis de
WG, induciendo una activación de citoquinas que determinan la degranulación
de polimorfo nucleares neutrófilos y liberación de radicales de oxígeno
y enzimas líticas así como mediadores inflamatorios tales como TNFa e interleuquinas
1 y 8, que inducen daño tisular, retención de neutrofilos dentro del espacio
intravascular y daño endotelial.
En la fase inicial
de la inflamación granulomatosa se produce un perfil de citoquinas, concordante
con una respuesta TH-1 polarizado con liberación de IFNg y TNFa, produciéndose granulomas compuestos por linfocitos
asociados a células de línea mielomonocítica alrededor de pequeños focos
de necrósis colágena. En la fase de generalización deWG se impone una
respuesta TH-2, y los granulomas están compuestos por una población celular
rica en linfocitos y eosinófilos.
Patología:
Las imágenes patológicas son variadas, pero entre los hallazgos característicos se incluyen:
Microabscesos neutrofílicos
y necrósis geográfica alrededor
de los cuales se disponen grupos de células gigantes multinucleadas hipercromáticas
y granulomas en empalizada ,con o sin vasculitis granulomatosa .
Estos hallazgos en presencia de test positivos
para PR3-ANCA, deberían ser diagnósticos pero formas limitadas a pulmòn
y tracto respiratorio superior, sólo muestran positividad en un 30 % de
los casos y en estas circunstancias una etiología infecciosa debe
ser cuidadosamente excluida.(
Fig.15 )
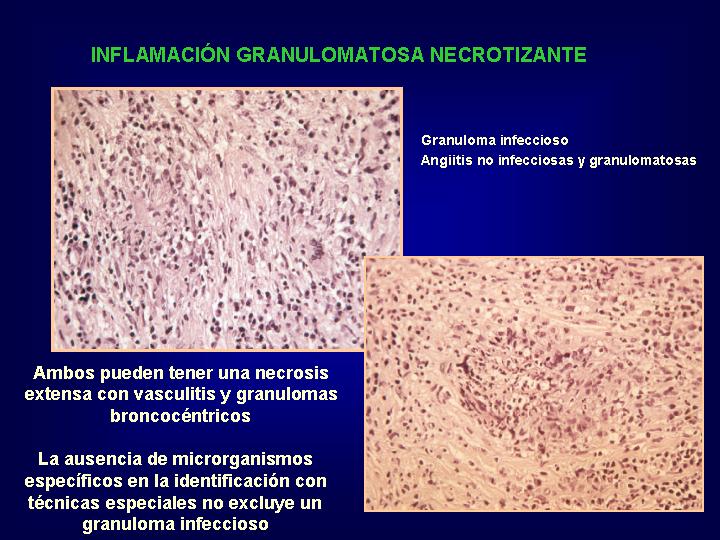{kind=link}
Los granulomas infecciosos
sueln presentar granulomas sarcoideos más allá de la zona de inflamación , la zona de necrosis
es redondeada y no muestra el aspecto irregular y geográfico del WG. Es
infrecuente la presencia de grupos de células gigantes multinucleadas
y hipercromáticas , así como la presencia de vascularitis segmentaria
fuera de la zona denecrosis.
La
granulomatosis de Wegener en el pulmón, evoluciona a traves de fases que
corresponden a enfermedad aguda, subaguda y crónica, estas fases pueden
ser subclasificadas como formas vasculares y extravasculares. Interpretando
el proceso como una enfermedad colágeno vascular que es una vasculitis
cuando hay necrosis e inflamación de los vasos sanguineos, bronquiolitis
cuando hay necrosis e inflamación de los bronquiolos o neumonía cuando
hay necrosis e inflamación histiocítica del parénquima.(Fig.
16 )
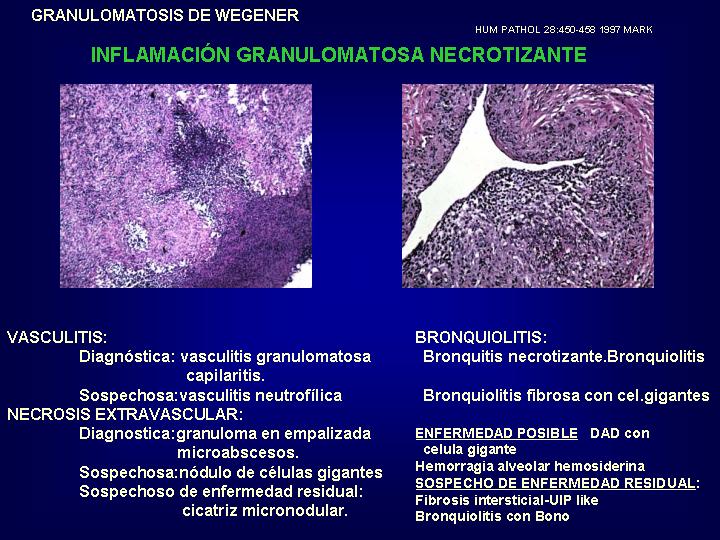{kind=link}
Diagnóstico diferencial:
- Reacciones a drogas:retinoides a-metil dopa y propil tiouracilo pueden inducir
cambios histológicos análogos a WG.
- Vasculitis granulomatosas:
Síndrome
de Churg-Strauss ( los pacientes tienen una historia de asma o enfermedad
estacional mediada por IgE.)
Granulomatosis
sarcoidea necrotizante ( hay grandes áreas de granulomas sarcoideos confluyentes)
·
Síndrome hemorrágico
alveolar producido por enfermedad colágeno vascular o enfermedad de Goodpasture.
Nódulos reumatoideos
necrobióticos (Fig.17
)
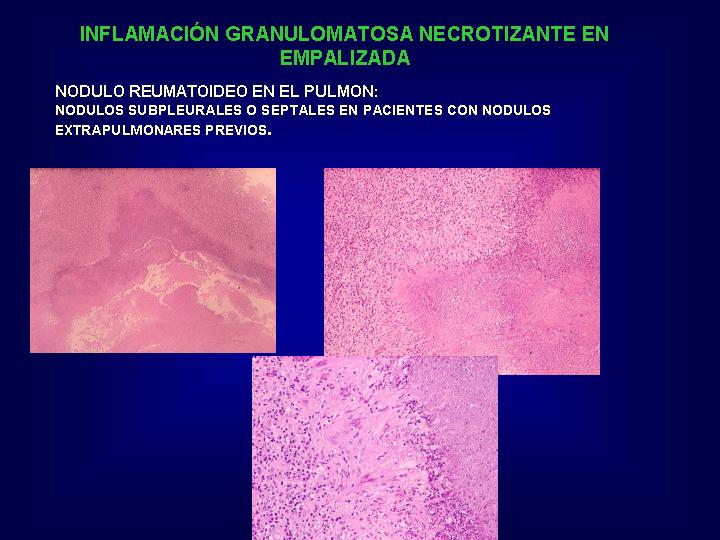{kind=link}
·
Granulomatosis linfomatoide:
Entidad
histopatológica caracterizada por infiltrado linfoide polimorfo y atípico
con prominente infiltración vascular ( angeitis ) y variable necrosis
(granulomatosis).
Los
análisis inmunohistoquímicos han demostrado que constituye la via común
de un grupo heterogéneo de desórdenes linfoproliferativos.
La
mayoría de los casos corresponde a un linfoma de células B clonales, infectadas
por EBV con una prominente respuesta de células T, al igual que en los
linfomas B ricos en células T reactivas.
Un
pequeño grupo contiene células T atipicas y carecen de células B atípicas,
son negativos para EBV y su comportamiento clínico agresivo sugiere un
linfoma T periférico.
Finalmente
los desordenes linfoproliferativos postrasplante y linfomas de células
T natural killer/extranodal tipo nasal son indistinguibles de la granulomatosis
linfomatoide cuando afectan al pulmón.
SINDROME
DE CHURG-STRAUSS
Vasculitis
sistémica de muy baja incidencia, en la cual el diagnóstico se establece
en biopsias extrapulmonares (preferentemente piel).
La
historia clínica y el patrón radiológico tiene un perfil típico.
Criterios
diágnosticos incluyen al menos cuatro de estos parámetros: a) asma, b)eosinofilia
en sangre periférica de más del 10 %, c) mono o polineuropatía, d) infiltrados
pulmonares transitorios, e) síntomas de senos paranasales, f) evidencia
histológica de infiltrados perivasculares eosinofílicos.
En
su fisiopatología se reconocen tres fases: pródromo asmático, eosinofilia
en sangre y tejidos y finalmente fase vasculítica.
Los
hallazgos histológicos en pulmón recuerdan una neumonía eosinofílica crónica,
con infiltrados alveolares e intersticiales, a los que se suman una vasculitis
eosinofílica de arterias capilares y venas. Las lesiones vasculiticas
contienen células gigantes y granulomas. (
Fig.18 )
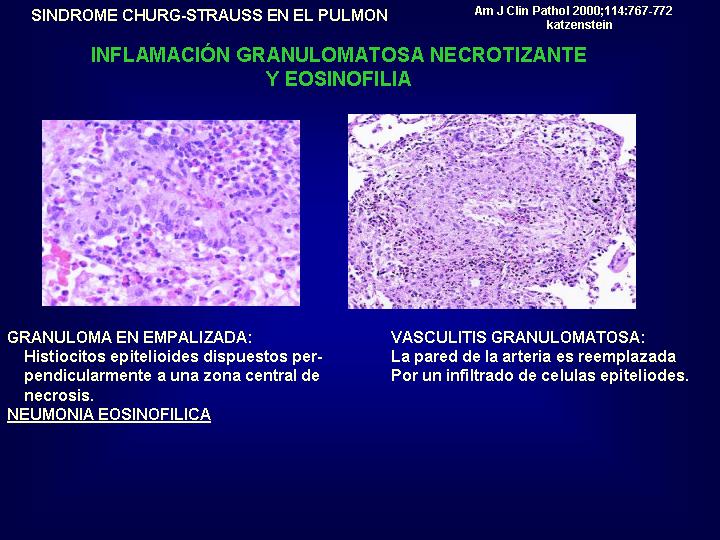{kind=link}
El
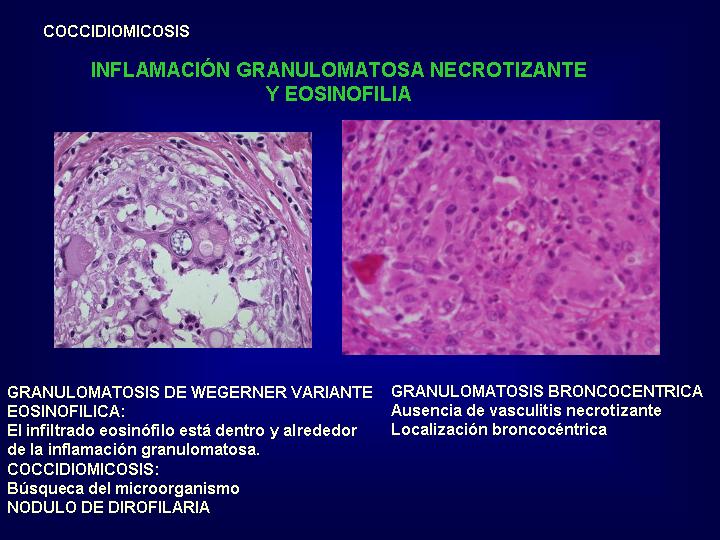
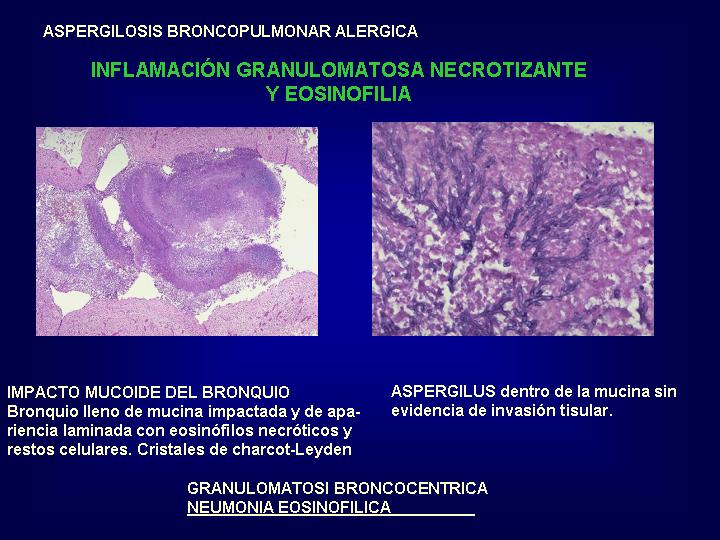
diagnóstico diferencial histológico se plantea con otras vasculitis, preferentemente
variante eosinofílica de granulomatosis de Wegener y con la neumonía eosinofílica
crónica y las entidades asociadas.( Fig.19 y 20
)
{kind=link}
{kind=link}
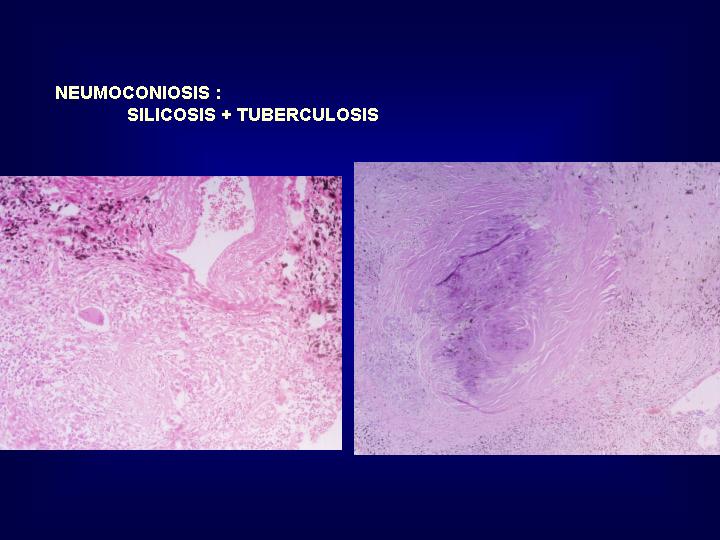
GRANULOMATOSIS HISTIOCITARIA
La mayoría corresponden
a neumoconiosis inducidas por polvo inorgánico. Las partículas
minerales son de una alta toxicidad que producen la rotura de membranas
lisosomales con una permanente activación y destrucción de macrófagos
que inducen fenómenos de fibrosis.Tienen una disposición preferentemente
peribronquiolar, constituyendo zonas hialinizadas rodeadas por macrófagos
y linfocitos. ( Fig. 21 )
{kind=link}
El paciente inmunosuprimido
es incapaz de mostrar una reacción de hipersensibilidad que determina el granuloma
sarcoideo, lo cual implica que en granulomatosis histiocitarias sin particulas
visibles deben realizarse técnicas especiales para excluir una etiología
infecciosa (Fig.22
)
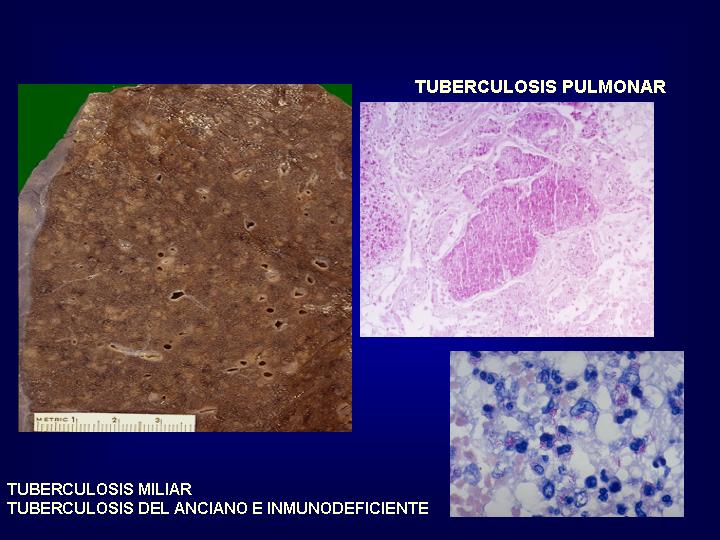{kind=link}
{kind=link}
![]() Conganat-
2002
Conganat-
2002
Uninet.edu & Club de Informática Aplicada de la SEAP
Correo-e contacto mailto:conganat@uninet.edu