|
Incidencia e impacto de las enfermedades neurodegenerativas
La reciente epidemia de encefalopatía espongiforme
bovina en el Reino Unido y su relación con la variante de la enfermedad
de Creutzfeld Jakob, que afecta de forma inusual a pacientes jóvenes,
ha suscitado un renovado interés por las enfermedades neurológicas. El
propio Prusiner, que recibió el premio Nobel por demostrar la capacidad
infectiva de la proteína PrP, comunica la relación de gran número de enfermedades
neurodegenerativas con trastornos de las proteínas, las cuales se acumulan
en el SNC bien porque se sinteticen de forma anómala, en exceso, cambien
su conformación o se dificulte su eliminación (Prusiner 2001).
En pocos años se han podido identificar
mutaciones o polimorfismos de los genes que regulan la síntesis de la
proteína b A4 (b
-amiloide), causante de la enfermedad de Alzheimer, de la a
-sinucleína, (enfermedad de Parkinson y otras a
-sinucleinopatías), de la TAU (complejo Pick, demencia frontal o frontotemporal,
parálisis supranuclear progresiva, etc.) SOD (esclerosis lateral amiotrófica),
huntingtina (enfermedad de Huntington), ataxina 1,2 y 3 (ataxias espinocerebelosas)
y proteína priónica. Uno de los principales campos de la investigación
de los patólogos es el estudio de las proteínas, la proteómica. Algunas
de las técnicas como la inmunohistoquímica están al alcance de la mayor
parte de los patólogos; Otras, como la electroforesis o la cromatografía
requieren manos expertas.
Aunque los mecanismos íntimos de muerte
neuronal, desarrollo y progresión de algunas de estas enfermedades continúan
siendo un enigma, se ha avanzado notablemente en los últimos años. Por
lo tanto hemos de tomar conciencia de que la contribución de los patólogos
al conocimiento de las enfermedades degenerativas del sistema nervioso
es, en el momento actual, imprescindible.
En España, a raíz de una iniciativa que
surge de patólogos y neurólogos, y amparada por el Ministerio de Ciencia
y Tecnología, se ha creado una red de Bancos de tejidos Neurológicos.
Requiere donación expresa de las muestras y su finalidad principal es
la investigación y el diagnóstico. Ello hace imprescindible que la obtención
de las muestras sea homogénea y los criterios neuropatológicos, actualizados.
Criterios neuropatológicos
Para el diagnóstico de estas enfermedades
es necesario la evaluación macroscópica del cerebro, el peso y describir
la existencia y distribución de atrofia en diferentes áreas corticales
y subcorticales. Se aconseja estudiar bloques de todos los lóbulos de
la corteza, hipocampo, ganglios basales, sustancia negra, cerebelo, tronco
y médula espinal. Las técnicas más frecuentemente utilizadas son H&E,
PAS, Plata metenamina, Luxol-Fast-Blue, Gallyas y Bielchowsky. Mediante
IHQ se usan anticuerpos contra la proteína TAU, b
-A4, ubicuitina , neurofilamentos, a -sinucleína,
y proteína acídica glial, etc.
a-SYNUCLEINOPATÍAS:
La familia de las sinucleínas está constituida
por tres miembros de los cuales a -sinucleína
y b -sinucleína son muy abundantes en el
sistema nervioso central y la g -sinucleína
en sistema nervioso periférico. Las funciones definidas de la a-sinucleína
son poco conocidas pero tiene funciones de chaperonina y es un regulador
de la actividad dopaminérgica dependiente de actividad. Esta proteína
es detectable mediante inmunohistoquímica y está presente también en las
placas seniles ya que el componente no amiloide de las placas contiene
en su molécula fragmentos del péptido precursor de la a-sinucleína.
Las a-synucleinopatías se caracterizan
por la formación de precipitados fibrilares de esta proteína. Son a-synucleinopatías:
a)la enfermedad de Parkinson, EP
b)la demencia con cuerpos de Lewy, DCL
c)la variante con cuerpos de Lewy de la EA ,vCLEA
d)la atrofia multisistémica en sus variantes
parkinsoniana AMS-p (más del 80% de los casos)
cerebelosa AMS-c (alrededor del 15%)
con disfunción autonómica, Shy-Drager
En todas ellas hay depósito de a-sinucleína
intracelular.
Los cuerpos de Lewy, cuyo componente proteico principal
es la a-sinucleína, se ven en la Enfermedad
de Parkinson en neuronas de la sustancia negra y núcleos pigmentados
y en la Demencia con cuerpos de Lewy DCL en neuronas corticales
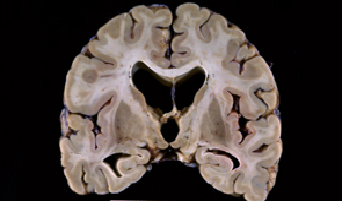
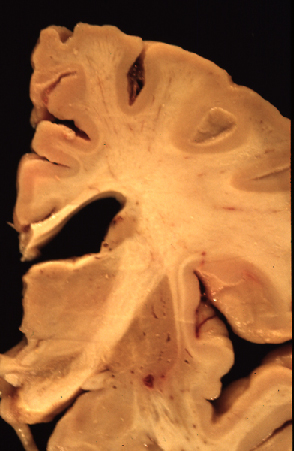
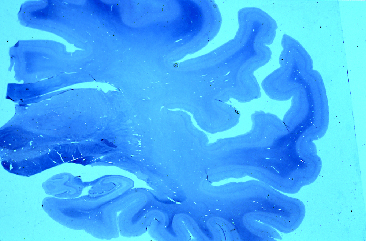
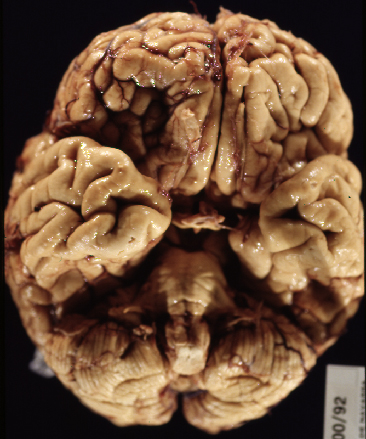
 Fig 1: Corte coronal de un cerebro con ECL que
presenta dilatación ventricular.
Fig 1: Corte coronal de un cerebro con ECL que
presenta dilatación ventricular. |
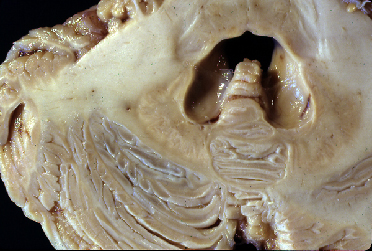
 Fig 2: Despigmentación del locus ceruleus y dilatación
característica del IV ventrículo
Fig 2: Despigmentación del locus ceruleus y dilatación
característica del IV ventrículo |
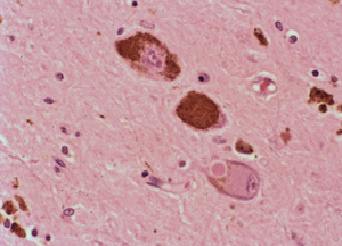
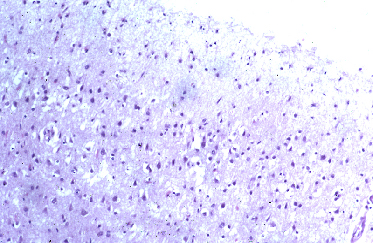
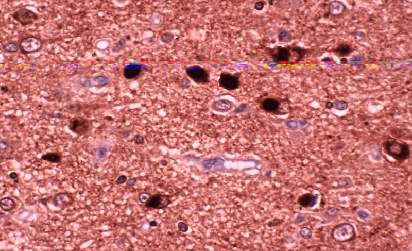
 Fig 3: Cuerpos de Lewy en neuronas de la sustancia
negra y microglía fagocitando melanina. H&E
Fig 3: Cuerpos de Lewy en neuronas de la sustancia
negra y microglía fagocitando melanina. H&E |
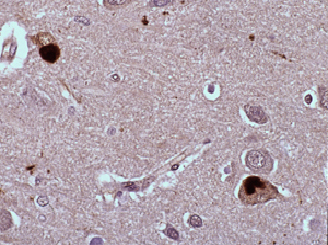
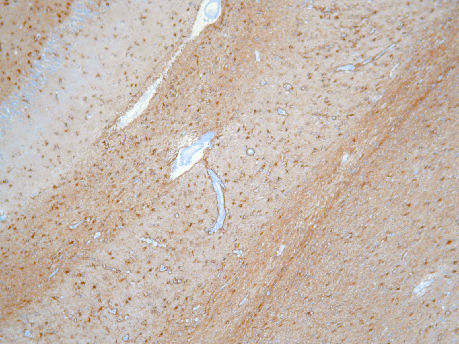
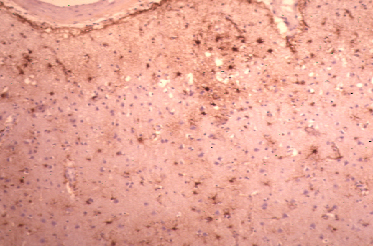
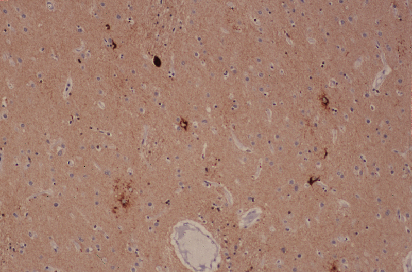
 Fig 4: Cuerpos de Lewy en neuronas de la corteza.
a-Sinucleína
Fig 4: Cuerpos de Lewy en neuronas de la corteza.
a-Sinucleína |
Se debe hacer cuantificación.
 Fig 5: La cuantificación de los cuerpos de Lewy se
debe hacer de 0 a 5 por área en varios campos de la corteza cerebral.
Fig 5: La cuantificación de los cuerpos de Lewy se
debe hacer de 0 a 5 por área en varios campos de la corteza cerebral.
Este tipo de demencia, más común de lo que
se sabía hasta hace pocos años, tiene dos formas anatomoclínicas, la forma
pura, DCL, que solamente presenta cuerpos de Lewy y la Variante con cuerpos
de Lewy de la enfermedad de Alzheimer, vCLEA, que asocia en la corteza
placas y ovillos.
Se conoce como Atrofia multisistémica, AMS,
una enfermedad neurogenerativa esporádica que presenta grados variables
de parkinsonismo, ataxia cerebelosa y disfunción autonómica que no suele
ir acompañada de deterioro cognitivo. En el cerebro hay atrofia variable
que puede afectar a diferentes vías o sistemas. Será más intensa en la
zona posterolateral del putamen y zona ventrolateral de la sustancia negra
en la variedad AMS-p (antes degeneración estrío-nígrica) y de oliva bulbar,
cerebelo, pedúnculos y protuberancia en la AMS-c (antes OPCA).
 Fig 6: Forma parkinsoniana de la atrofia multisistémica.
Se observa aumento de coloración del estriado debido a la presencia
de pigmentos sobretodo hierro.
Fig 6: Forma parkinsoniana de la atrofia multisistémica.
Se observa aumento de coloración del estriado debido a la presencia
de pigmentos sobretodo hierro. |
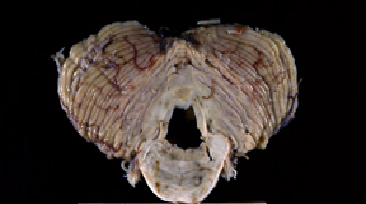
 Fig 7: Forma cerebelosa de la atrofia multisistémica:
adelgazamiento de la corteza cerebelosa, de los pedúnculos, coloración
grisácea de la sustancia blanca del hilio del dentado, afectación
pontina y dilatación del IV ventrículo.
Fig 7: Forma cerebelosa de la atrofia multisistémica:
adelgazamiento de la corteza cerebelosa, de los pedúnculos, coloración
grisácea de la sustancia blanca del hilio del dentado, afectación
pontina y dilatación del IV ventrículo. |
Microscópicamente, hay pérdida neuronal
es las zonas afectadas y depósito de hierro en el putamen. Pero la clave
diagnóstica de es la presencia de inclusiones intracitoplasmáticas en
oligodendroglía, microglía, astrocitos y neuronas, que son inmunoreactivas
con TAU, aB-cristalina y a-sinucleína.
La oligodendroglía parece que juega un papel determinante en el desarrollo
y progresión de la enfermedad.
ENFERMEDAD DE ALZHEIMER
El diagnóstico clínico-patológico de la
enfermedad de Alzheimer, EA, se lleva a cabo siguiendo los criterios CERAD
que tiene en cuenta los factores de edad y densidad de placas, (b-A4
+), y ovillos neurofibrilares (TAU+).
(Fig 8-14).
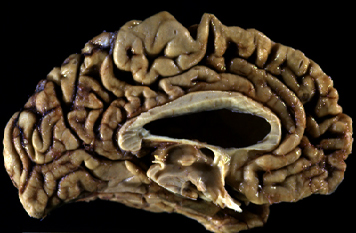
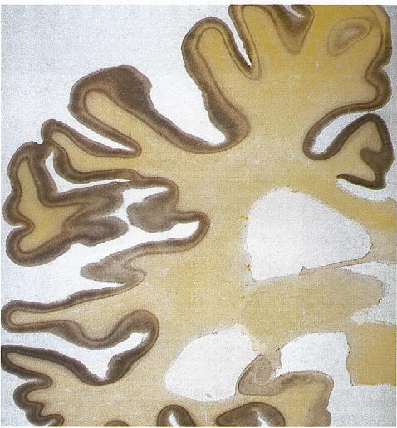
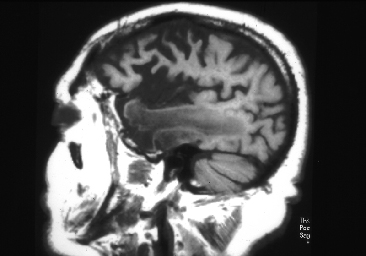
 Fig 8: Enfermedad de Alzheimer, EA, corte sagital
medial: surcos profundos y circunvoluciones adelgazadas en todos
los lóbulos.
Fig 8: Enfermedad de Alzheimer, EA, corte sagital
medial: surcos profundos y circunvoluciones adelgazadas en todos
los lóbulos. |

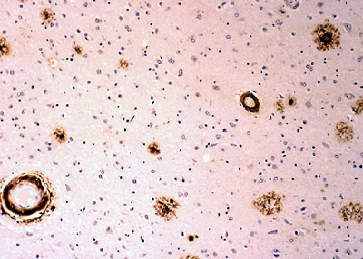
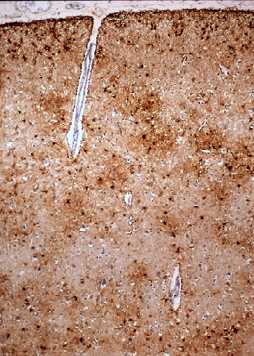
Fig 9: Corteza cerebral en la EA: tinción inmunohistoquímica
para la proteína b-amiloide que se
deposita en las placas seniles y en las paredes vasculares. |

Fig 10 : Corteza cerebral en la EA: tinción inmunohistoquímica
para la proteína glial acídica que demuestra la gliosis por pérdida
neuronal. |

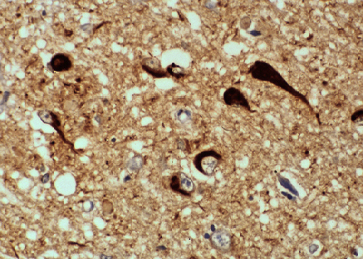
Fig 11 : Corteza cerebral en la EA: tinción inmunohistoquímica
para la proteína TAU, que se deposita en los ovillos neurofibrilares |

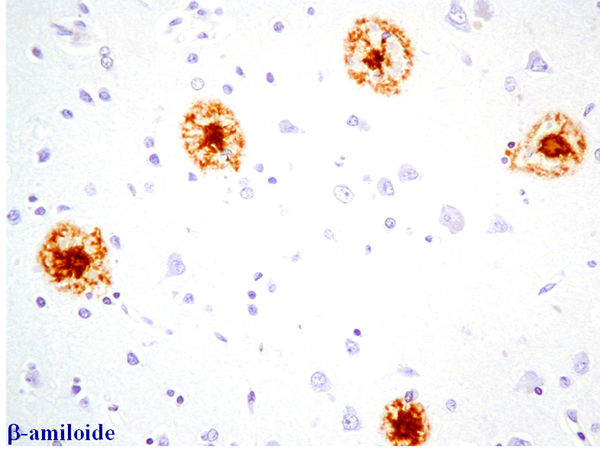
Fig 12: Corteza cerebral en la EA: tinción inmunohistoquímica
para la proteína b-amiloide en una
densidad de placas B. |

 Fig 13 y 14: Tablas de diagnóstico de EA. El diagnóstico
clínico, será de probable o posible, y será seguro únicamente si se hace
el estudio neuropatológico. La densidad de placas y ovillos se cuantifica
de menos a más en A,B y C.
Fig 13 y 14: Tablas de diagnóstico de EA. El diagnóstico
clínico, será de probable o posible, y será seguro únicamente si se hace
el estudio neuropatológico. La densidad de placas y ovillos se cuantifica
de menos a más en A,B y C.
El estadiaje se realiza según las recomendaciones de Braak y Braak (Fig
15 y 16).

 Fig 15 y 16: Estadios neuropatológicos según Braak
y Braak. Cortes hemisféricos coronales de dos cerebros con EA que demuestran
la densidad y distribución de los ovillos en estadios intermedios y avanzados
respectivamente de la enfermedad.
Fig 15 y 16: Estadios neuropatológicos según Braak
y Braak. Cortes hemisféricos coronales de dos cerebros con EA que demuestran
la densidad y distribución de los ovillos en estadios intermedios y avanzados
respectivamente de la enfermedad.
En las formas familiares de EA los genes
más habitualmente implicados son el gen de la proteína precursora del
amiloide (bAPP), ApoE4, presenilina 1 (PS1)
y presenilina 2 (PS2). La influencia de los factores vasculares en la
EA es admitida por todos. Los niveles elevados de homocisteína en plasma,
parece ser que contribuyen al desarrollo de la enfermedad favoreciendo
no solamente los factores de riesgo vascular sino también provocando la
muerte neuronal.
DEMENCIA VASCULAR
La causa de la demencia vascular puede tener
un substrato morfológico muy variable, lo cual permite clasificarla según
la Fig 17. No obstante, no es inhabitual que se asocien en el mismo cerebro
cambios vasculares con placas y ovillos (u otra patología degenerativa)
denominándose demencia mixta. El protocolo CERAD, aunque diseñado en un
principio para la EA es adecuado para describir las lesiones vasculares.
En las Figs 18-21 podemos ver diferentes lesiones que son causa de demencia.
La esclerosis isquémica del hipocampo, produce trastornos de memoria aunque
no siempre son progresivos (Fig 22).
 Fig 17: Clasificación de la demencia vascular
Fig 17: Clasificación de la demencia vascular

Fig 18: Demencia vascular debido a microinfartos
corticales en una paciente hipertensa.
| 
Fig 19 Demencia vascular: Leucoaraiosis e un caso
de angiopatía cerebral amiloidea |

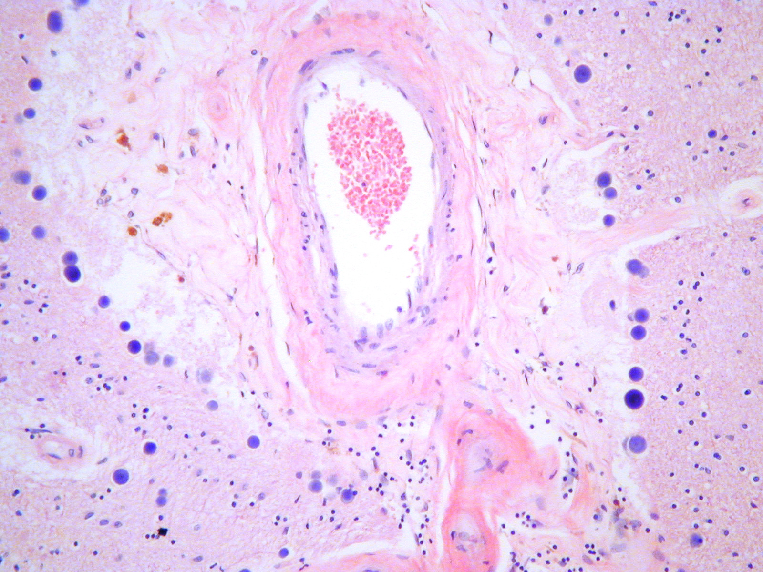
Fig 20: Demencia vascular: Infarto en la proximidad
del núcleo basal de Meynert. |

Fig 21: Demencia vascular: Microinfartos en sustancia
blanca en un caso de encefalopatía de Bienswanger. Tinción de Luxol-fast-blue. |

Fig 22: Demencia vascular: esclerosis isquémica
del hipocampo. Tinción inmunohistoquímica para la proteína glial que
demuestra la pérdida neuronal en el sector de Sommers. |
Hay un tipo de arteriopatía cerebral, CADASIL,
con herencia autosómica dominante, caracterizada por migraña e infartos
múltiples y demencia que comienza en la cuarta o quinta década. Más del
95% de los casos se asocian a mutaciones puntuales en el dominio extracelular
del gen Notch3. Este gen forma parte de la familia Notch que tiene 4 miembros
y mucha importancia durante el desarrollo. El Notch3 tiene que ver con
la organogénesis, concretamente con la miogénesis, controlando las proteínas
de las fibras musculares. Esto es importante porque en algunos casos permite
el diagnóstico por biopsia de piel, músculo o nervio, en donde se identifican
con ME inclusiones osmiófilas en las arteriolas dérmicas. En cerebros
de autopsia, se observan también estas inclusiones y con histoquímica,
depósitos Pas +, Rojo Congo - en la capa muscular de las arteriolas e
infartos lacunares múltiples tanto en corteza como en sustancia blanca.
(Fig 23).
 Fig 23: Demencia vascular: Enfermedad de CADASIL.
Fig 23: Demencia vascular: Enfermedad de CADASIL.
Los vasos de la sustancia blanca cerebral presentan depósitos proteináceos
que se tiñen con Pas.DEMENCIA CON GRANOS ARGIRÓFILOS
Es una patología infrecuente y mal conocida
por muchos neuropatólogos. Se define por la presencia de granos y bastones
en el neuropilo de la corteza cerebral que se ponen de manifiesto con
plata de Gallyas y expresan TAU y a -B
cristalina. Pero estudios recientes de la clínica Mayo no demuestran una
correlación con la presencia o no de trastorno cognitivo (Togo 2002).
Es independiente del genotipo apoE.
ENFERMEDADES POR PRIONES
Prion es el término acuñado por el Premio
Nobel de Medicina 1997 Prusiner, que significa "sólo proteína". Los priones
son partículas protéicas infecciosas que se diferencian de virus y viroides
por las siguientes características: carecen de ácidos nucleicos, no se
destruyen por ebullición (lo hacen a 133°C a una presión de 3 atmósferas
durante 20 minutos), no se destruyen con los desinfectantes habituales
(sí con el ácido fórmico), son muy resistentes a la descomposición biológica,
pueden sobrevivir en el suelo durante muchos años, son resistentes a las
proteasas, no provocan respuesta inflamatoria ni inmune y su diana es
el sistema nervioso central. Infectan por contacto y provocan un efecto
destructivo de las neuronas que se acelera progresivamente conocido como
"efecto dominó". En los diferentes tejidos del organismo, existe una proteína
priónica normal también llamada proteína priónica celular (PrPc) cuya
concentración es máxima en el SNC y linfocitos B y su función es desconocida.
La clasificación actual de las EETH. incluye:
formas esporádicas (ECJ: clásica, variantes clínicas, etc.), adquiridas
(Kuru, ECJ yatrogénica y la variante británica, vECJ) y familiares (ECJ
familiar, síndrome de Gerstmann-Straussler-Scheinker, insomnio familiar
letal y mutaciones atípicas).
El 85 % de los casos corresponden a la forma
clásica y esporádica de la ECJ y en mayor o menor medida todas presentan
espongiosis y expresan la proteína PrP. Hay distribución variable en el
tejido de la proteína y algunas diferencias morfológicas que conviene
reseñar.
a) Forma clásica o esporádica de CJ: Hay
atrofia de localización variable según las formas clínicas y pigmentación
ocre de la corteza cerebral y cerebelosa debido a la lipofuscina. El
hipocampo, a diferencia de otras demencias está respetado. Excepto en
la forma "panencefalopática" la sustancia blanca no está especialmente
dañada. Hay espongiosis, gliosis, y pérdida neuronal. La espongiosis
en la ECJ suele ser transcortical o bien de capas profundas (no laminar
ni superficial). Las vacuolas están en el neuropilo y en los somas neuronales
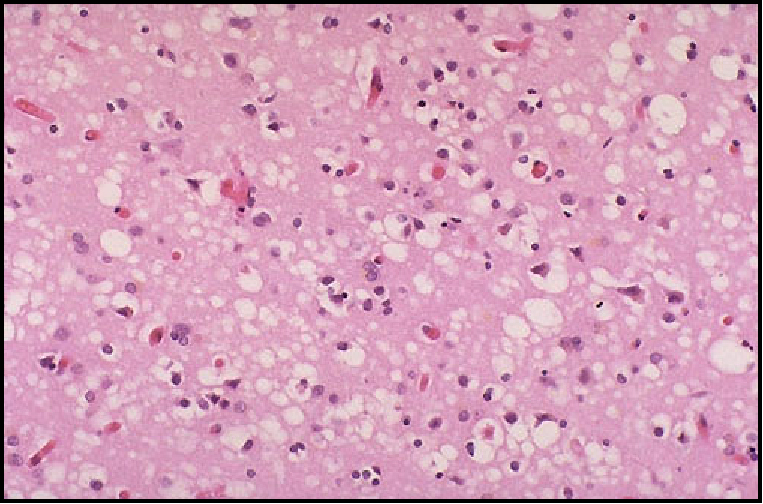
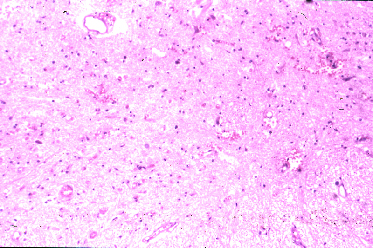
y el tamaño oscila entre 20 y 200 micras (Fig 24).
 Fig 24: EETH, Espongiosis en el neuropilo e intraneuronal.
H&E.
Fig 24: EETH, Espongiosis en el neuropilo e intraneuronal.
H&E.
El depósito de PrPsc en el tejido es clave
en el diagnóstico. Esto se puede hacer mediante inmunohistoquímica tras
digestión con proteinasa K (la PrPc desaparecerá y la PrPsc persiste)
o bien por Western-blot. Además hay depósitos de b-amiloide
por reactividad cruzada lo cual exige rigor en la metodología pues puede
plantear problemas diagnósticos con la enfermedad de Alzheimer. La distribución
de la proteína priónica es en placas, en vacuolas y en sinapsis.
b) Variante británica (vCJ): En estas
formas la neuropatología difiere bastante de las formas esporádias.
Los cambios espongiformes son casi inapreciables en la corteza y muy
claros en los ganglios de la base. En la corteza y de forma muy manifiesta
en la corteza cerebelosa hay un tipo peculiar de placas llamadas placas
floridas que recuerdan a estructuras florales, constituídas por un nucleo
denso de proteína PrP resistente a la proteinasa K, rodeado de vacuolas.
En núcleos talámicos posteriores y mesencéfalo (sustancia gris periacueductal),
astrocitosis marcada y pérdida neuronal; acúmulo masivo de PrPsc en
placas, perineuronal y perivascular. La despoblación neuronal de a corteza,
como en las formas esporádicas tiende a respetar el hipocampo. EL depósito
de la PrP en amígdala y otros tejidos linfoides ha quedado demostrado
y tiene valor diagnóstico. Se ha demostrado en el estudio retrospectivo
del apéndice de un paciente con una apendicitis operado seis meses antes
de aparecer el cuadro neurológico.
c) Gerstmann-Straussler- Scheinker (GSS):
Si el diagnóstico diferencial morfológico entre a y b es relativamente
fácil, no ocurre así entre las formas familiares de ECJ y GSS. Ambas
son tan similares que solamente recurriendo el estudio genético se pueden
diferenciar. Morfológicamente hay numerosas espongiosis y placas de
PrP proteinasa K resistente. Se han descrito además ovillos neurofibrilares
y degeneración de la sustancia negra.
d) Insomnio familiar fatal: Esta enfermedad
tan infrecuente, (de la cual hay un grupo de casos en España localizados
en el País Vasco, Navarra y Cantabria) se caracteriza por presentar
una clínica de demencia de corta evolución y trastornos graves del sueño,
(somnolencia de día e insomnio de noche). Tiene carácter familiar y
anticipación en el tiempo de una generación a otra. Se observan lesiones
talámicas y en las olivas bulbares (gliosis y pérdida neuronal, sin
espongiosis apenas).
La normativa legal y los cuidados que se
han de tomar en la realización de las autopsias con esta patología han
sido recogidos en un documento reciente. ( http://www.eeb.es) TAUPATÍAS
En los últimos años se discute quién define
si las enfermedades deben definirse por los síntomas clínicos, por los
cambios histológicos o por la genética. Uno de los exponentes más significativos
ocurre en neurociencias desde el conocimiento de la patología tau.
 Fig 25: Clasificación de las taupatías (Mckhan 2001)
Fig 25: Clasificación de las taupatías (Mckhan 2001)
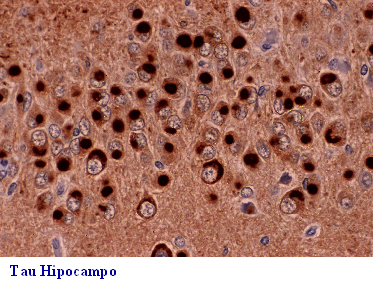
La proteína TAU pertenece a la familia de
las proteínas asociadas a los microtúbulos. Su función es el ensamblaje
y estabilización de los microtúbulos que son organelas vitales en la formación
de las neuritas y en el transporte intraneuronal.
Está codificada por un único gen localizado en el brazo largo del cromosoma
17 (17q21); este gen tiene 14 exones y del se producen, mediante el
mecanismo conocido como splicing alternativo, 6 mRNAs diferentes
que dan lugar a 6 isoformas de la proteína. Las diferentes isoformas
de TAU difieren entre sí en la presencia o ausencia de las secuencias
peptídicas codificadas por los exones 2, 3 y 10. Tres isoformas de TAU
contienen la secuencia polipeptídica correspondiente al exón 10 (10+)
también denominada 4R porque contienen cuatro secuencias repetitivas
insertadas y otras tres isoformas son 10- porque no han traducido el
exón 10 y se denominan 3R porque solo tienen 3 secuencias repetitivas.
Estas variaciones son importantes desde
el punto de vista funcional porque dependiendo de que sean isoformas 10+
ó 10-, tienen 4 ó 3 puntos de anclaje con los microtúbulos. En el cortex
cerebral normal se expresan tanto las formas 3R como las 4R pero de una
forma diferenciada en las distintas subpoblaciones neuronales lo que indica
que, la distinta conformación proteica tiene una función fisiológica determinada.
Además del número de puntos de anclaje con
los microtúbulos, en la funcionalidad de la proteína TAU, tiene gran importancia
el estado de fosforilación de la misma: las proteínas hiperfosforiladas
son menos eficientes que las no fosforiladas. El estado de hiperfosforilación
las hace más largas y rígidas lo que facilita su agregación y depósito
en forma de inclusiones filamentosas. De hecho, es una evidencia que en
la enfermedad de Alzheimer (EA) la degeneración neurofibrilar está constituida
mayoritariamente por proteína TAU hiperfosforilada.
Desde 1994, se conocen familias con clínica
de demencia fronto-temporal (DFT) con rasgo de herencia autosómica dominante.
En aproximadamente la mitad de estos casos estos casos se ha podido demostrar
alguna mutación del gen TAU. Estos casos, bastante heterogéneos desde
el punto de vista clínico, se han agrupado bajo el término de FTDP-17
(fronto temporal demencia y parkinsonismo asociado al Cr 17). Esta es
la única entidad con mutaciones del gen TAU, de lo que se deduce que en
el resto de DFT claramente familiares otros genes deben estar implicados.
Todas las taupatías tienen en común además
de depósito en cerebro de formas anormales de la proteína tau, la atrofia
lobar, un mayor o menor grado de vacuolización cortical superficial, gliosis
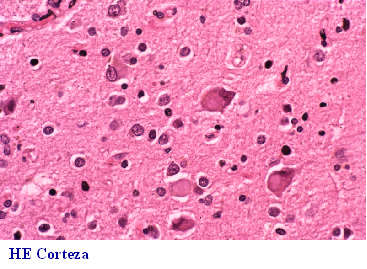
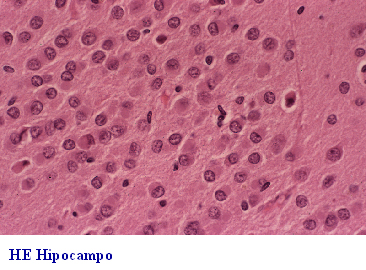
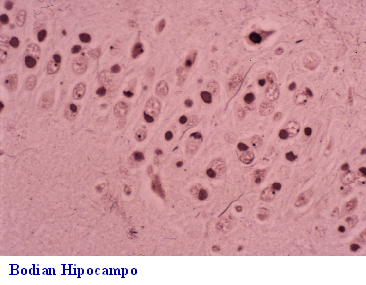
subcortical y neuronas abalonadas. La demencia frontal o frontotemporal
DF-DFT, (Fig 26-36), además de afectación lobar y pérdida neuronal, puede
presentar dos formas anatomopatológicas: a) con neuronas argirófilas y
abalonadas de Pick (Fig 29)y cuerpos de Pick de localización en la capa
granular del hipocampo(Fig 30-32) y b) sin cuerpos de Pick; esta última
forma, puede cursar con una clínica frontal muy manifiesta que contrasta
con la escasa expresión morfológica que en ocasiones ha sido denominada
Demencia frontal sin cambios y que muestra desmielinización subcortical
espongiosis y gliosis de capas superficiales y a veces se asocia a pérdida
de neuronas en el asta anterior de la médula espinal (Fig 33-36). La Parálisis
supranuclear progresiva, PSP, microscópicamente presenta atrofia del tronco
(Fig 37), y las neuronas que expresan tau están sobretodo en ganglios
de la base, tronco del encéfalo y pars compacta de la sustancia negra
(Fig 38 y 39); destaca también la presencia de astrocitos tau + en estas
zonas sobretodo en el putamen. La Degeneración cortico-basal-gangliónica
CBG, asocia la presencia de neuronas abalonadas, acromáticas, (se diferencian
de los cuerpos de Pick por la ausencia de argirofilia) en corteza parietal
y placas astrocitarias características que expresan proteína acídica glial
PGAF y tau.
ENFERMEDAD DE HUNTINGTON
Esta forma de corea se expresa clínicamente
con movimientos atetósicos, trastornos psiquiátricos y demencia; es debida
a una secuencia repetida de tripletes de bases GAG en el gen IT15 que
codifica una proteína denominada Huntingtina. Tiene una herencia autosómica
dominante, comienza en la cuarta o quinta década y presenta el fenómeno
de anticipación (en una misma familia la edad en la generación siguiente
es menor). Existen varios modelos experimentales en los cuales la hipótesis
excitotóxica es la más convincente. El estudio neuropatológico demuestra
que la diana en esta enfermedad es las neuronas gabaérgicas del núcleo
estriado con aumento del glutamato y disfunción el equilibrio entre el
estriado y la corteza afectando a la vía directa D1 y a la vía indirecta
D2. Microscópicamente destaca la atrofia del estriado sobretodo de la
cabeza del núcleo caudado (Fig 40). Se demuestran inclusiones intranucleares
y neuritas distróficas que expresan Huntingtina y ubicuitina.
 Fig 40: Enfermedad de Huntington mostrando marcada
atrofia del núcleo caudado y dilatación ventricular.
Fig 40: Enfermedad de Huntington mostrando marcada
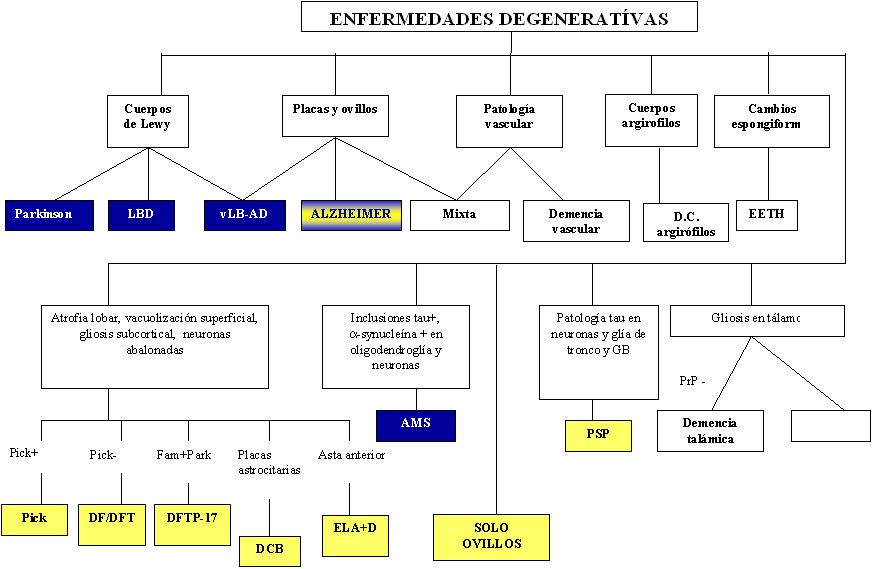
atrofia del núcleo caudado y dilatación ventricular. En la Figura 41 presentamos un algoritmo diagnóstico en las enfermedades
neurodegenerativas.
 Fig 41: Algoritmo diagnóstico de las enfermedades neurodegenerativas.
Fig 41: Algoritmo diagnóstico de las enfermedades neurodegenerativas.
Bibliografía
Neuropathological diagnostic criteria for brain banking.
F.F. Cruz-Sanchez. R David y ML Cuzner (eds). IOS press. Amsterdam 1995.
Panizo-Santos A, Roberts D, Merino MJ. Proteomic applications (II).
Rev Esp Patol 2001; 34: 177-187.
Graeber MB and Morán LB. Mechanism of cell death in neurodegenerative
diseases: fashion, fiction and facts. Brain Pathol 2002; 12:385-390.
Criterios diagnósticos neuropatológicos
a-sinucleínopatías
Hashimoto M. Brain Pathol 707-720,1999
Ferrer I. Neurología 16:163-170, 2001.
Dickson DW, Lin W, Liu W and Yen SH. Brain Pathol 1999; 9: 721-732
Burns DJ. J. Clin Pathol:Mol Pathol 2001; 54:419-426.
Enfermedad de Alzheimer
McKhannG. Neurology 34:939-944,1984 (NINCDS/ADRDA)
Braak H . Acta Neuropathol 82: 239-259, 1991 (estadiaje)
Mirra SS. Neurology 41:479-486,1991. (CERAD)
Heyman A. Neurology 52:1839-1844,1999 (CERAD XIX:variante con cuerpos
de Lewy)
Loscalzo J NEJM 346:476-485, 2002. (factores vasculares-homocistinemia)
Parkinson
Mirra SS. Neurology 41:479-486,1991. (CERAD)
Hulette C. Neurology 45:1991-1995, 1995 (CERAD IX: Parkinson y Alzheimer)
E.Masliah, Brain Pathol : 707-720,1999
Shimura H Science 13,293(5528):263-269,2001
Demencia con cuerpos de Lewy
Mc Keith. Neurology 47:1113-1124, 1996.
Heyman A. Neurology 52:1839-1844,1999 (CERAD XIX: Alzheimer con cuerpos
de Lewy)
Demencia vascular
Roman GC. Neurology 43: 250-260. 1993;
Kalimo H . Brain Pathol 12:371-384, 2002;
Demencia con cuerpos argirófilos
Jellinger KA. Brain Pathol 8:377-386,1998.
Togo T Brain Pathol 2002; 12:45-52, 2002.
Enfermedades por priones
Budka H. Brain Pathol 5: 459-566,1995.
Parchi P. Ann Neurol 46:224-233, 1999
Ironside JW. Histopathology 37:1-9, 2000
Prusiner SB.N Eng J Med, 2001 Vol 344, 20: 1516-1526. (http://www.nejm.org)
T.Tuñón. Libro de ponencias del XX congreso nacional de la SEAP. Julio.
ISBN 84-699-5285-4. 2001
Budka H. Brain Pathol 12:1-11, 2002
Doc Bioseguridad . Opinión CMIEET 2002 (http://www.eeb.es)
Taupatías
Komori T. Brain Pathol 9:663-679,1999
Tolnay M. Neuropath Appl Neurobiol 26: 368-378,2000
McKhan GM Arch Neurol 58:1803-1809. 2001;
Morris HR Arch Neurol; 58:1813-1816.2001
Demencia frontal o frontotemporal
Kertesz A. Arch Neurol 55:302-324,1998
Mann DMA. Brain Pathol 8:325-338,1998.
Dickson DW. Brain Pathol 8:339-354,1998.(enfermedad de Pick)
Spillantini MG. Brain Pathol 8:387-402,1998 (FTDP-17)
J M Manubens, T.Tuñón . ed S.L. Pousa .Prous Science Barcelona 2001
163-181.
Nakano I Neuropatholgy 1:68-75,2000 (asociada a enfermedad de motoneurona)
Degeneración corticobasal
Riley DE. Neurology 40:1203-1212,1990
Bergeron C. Brain Pathol 8:355-365, 1998 (con demencia)
Kertesz A. Neurology 55:1368-1375,2000 (relación con la afasia progresiva
primaria y la demencia frontal)
Parálisis supranuclear progresiva
Bergeron C. Brain Pathol 8:355-365, 1998 (con demencia)
Tawana K J. Clin Pathol: Mol Pathol 2001; 54:427-434.
Enfermedad de Huntington
Greenfield, Vol 2 , pag 305 y ss 1997
Vonsatell and DiFligia. J: Neuropathol Exp Neurol 57(5): 369-384,
1998.
Schieman M J Neuropathol Exp Neurol 58(2):129-137,1999.
|