| IV CONGRESO VIRTUAL HISPANO AMERICANO DE ANATOMÍA PATOLÓGICA |
|
CONTENIDO |
|
|
|
|
|
IMÁGENES | ||||||||||||||||||
|
|
|
|
INTRODUCCIÓN | |
|
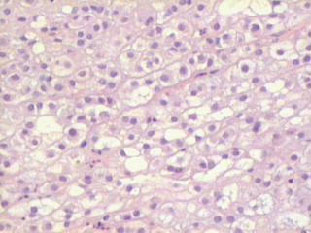
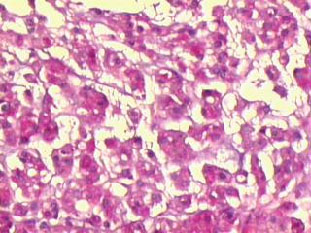
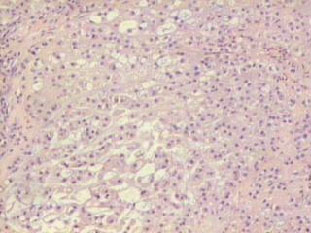
El cordoma es una neoplasia maligna que se origina de restos o vestigios de la notocorda . Esta estructura embrionaria es el resultado de la invaginación de células prenotocordales en la región de la fosa primitiva, las mismas que emigran directamente en dirección cefálica hasta llegar a la lámina precordal. Estas células se intercalan en el hipoblasto, de manera que durante un período breve, la línea media del embrión está formada por dos capas celulares que constituyen la lámina notocordal, luego se desprenden del endodermo, formando un cordón macizo, llamado notocorda definitiva que se encuentra por debajo del tubo neural y sirve de base para el esqueleto axial, de tal manera que esta estructura se extiende desde la lámina precordal (la futura membrana bucofaríngea) hasta la zona caudal en relación con la fosa primitiva. Lo anteriormente mencionado explica el porque este tumor se puede encontrar dentro de los huesos del cráneo, en la unión esfenooccipital y en dorsum sellae. También se han encontrado ectopias notocordales en la porción lateral y ventral del puente (1). La gran mayoría de los cordomas se originan en la región sacrocoxígea, en donde invaden, erosionan y destruyen el hueso, así como estructuras vecinas. Aproximadamente el 60% de los cordomas afectan las vértebras en esta localización. Los tumores intracraneanos constituyen un 40% y ocurren en dos localizaciones predominantemente: la más común es la región del clivus o la unión esfenooccipital y la otra es en la vecindad de la región selar. Cuando la lesión se localiza en el clivus los síntomas más frecuentes son alteraciones visuales y trastornos relacionados con la marcha. Es un tumor poco frecuente , constituye el 4% de todos los tumores malignos primarios del hueso. La edad de afección varía entre los 40 y 80 años es raro observarlo antes de los 30 años y cuando esto sucede la localización más frecuente es a nivel esfenooccipital. El sexo masculino es el más afectado en una proporción 2:1. La localización según la frecuencia es : sacrocoxígea 50% , esfenooccipital 37%, región cervical 6%, región lumbar 4%, región torácica. Macroscópicamente son tumores multilobulados de color gris rojizo, con zonas traslúcidas y consistencia firme, puede haber hemorragias focales y calcificaciones. Estas neoplasias crecen generalmente hacia la línea media destruyen, invaden huesos, comprimen estructuras nerviosas vitales, frecuentemente se alojan en el ángulo pontocerebelosos produciendo su deformación, puede llegar hasta el seno cavernoso y esfenoidal y cuando su extensión es en sentido antero inferior se presenta como masa nasofaríngea. Se han observado cordomas que por su tamaño produce exoftalmos. Microscópicamente este tumor se dispone en nidos, lóbulos, o cordones de células claras, rodeados por un estroma mixoide o conectivo. La morfología celular es variable, predomina el grado de vacuolización que presenta el citoplasma, puede haber pocas o múltiples vacuolas , de tamaño grande o pequeño que desplazan el núcleo (células fisalíferas) y contienen material mucinoso (glucógeno) intra o extracelular que es positivo para la tinción de PAS o Mucicarmín. Los núcleos presentan alguna variación en el tamaño y pueden verse algunas mitosis atípicas. Estas formas celulares posiblemente representan diferentes estadíos de diferenciación de la notocorda que es de origen ectodérmico, sin embargo puede haber diferenciación condroide o contener hueso por formación o invasión del mismo. En el diagnóstico diferencial, por las características histológicas , otras neoplasias deben ser descartadas, entre ellas el condrosarcoma, metástasis de carcinoma de células claras del riñón o célula en anillo de sello, por esta razón es necesario recurrir a la histoquímica y sobre todo a la inmunohistoquímica (citoqueratinas, EMA, y lo más importante la proteína S-100 que es intensamente positiva y traduce su origen neuroectodérmico). Inmunohistoquímicamente la demostración de marcadores epiteliales es útil para distinguir de los condrosarcomas (2, 4).
|
||
|
|
MATERIAL Y MÉTODOS - RESULTADOS | |
|
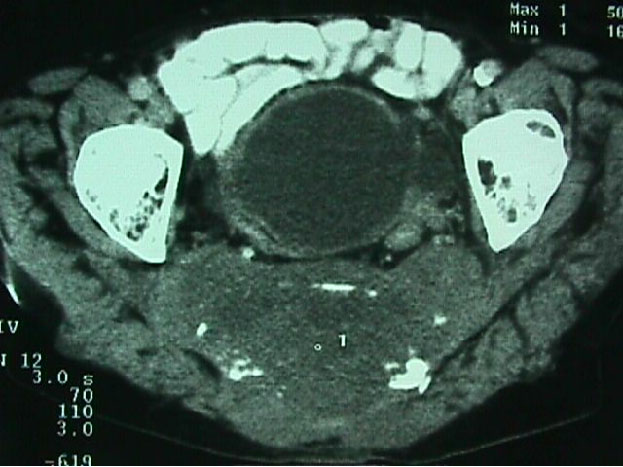
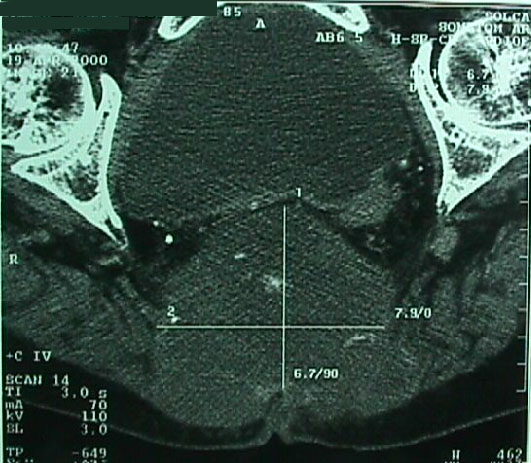
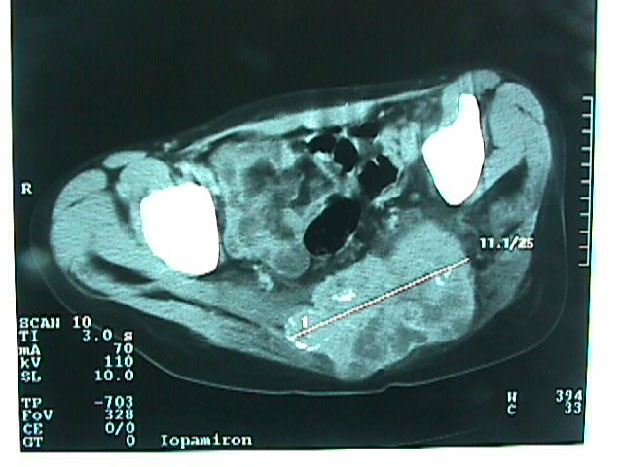
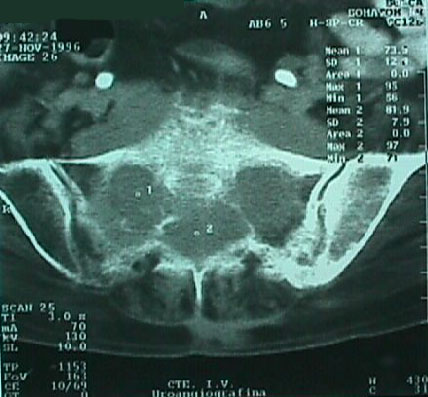
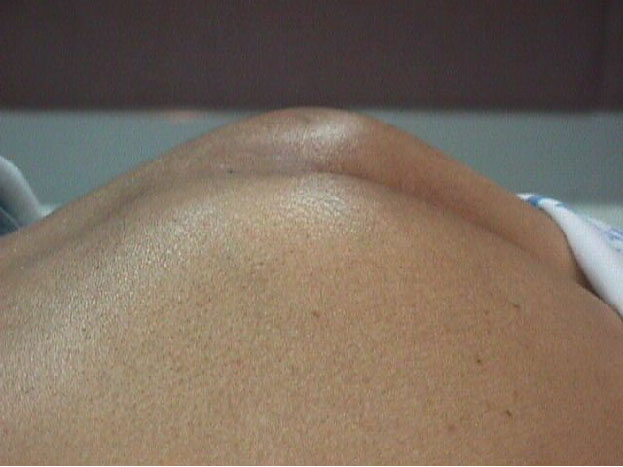
Caso # 1: Paciente femenina de 85 años de edad procedente de Girón , ocupación QQDD, que desde hace unos 8 meses presenta dolor pélvico y nota la presencia de dos tumoraciones en región glútea que ha aumentando progresivamente de tamaño, actualmente tiene un diámetro de 15 cm. Se acompaña de incontinencia urinaria y dolor a la defecación. El examen físico pone en evidencia una lesión de aproximadamente 15 cm de diámetro, localizada a nivel de la región sacra, intensamente dolorosa y de bordes irregulares (Figura 1). En la TAC se observa lesión ocupativa que compromete de manera difusa toda la región correspondiente al sacro en su porción inferior con estallamiento y destrucción del mismo, existe componente de tejidos blandos hacia los agujeros ciáticos mayores y menores (Figura 2) y (Figura 3). Histopatología: Esta lesión presenta infiltración de los tejidos blandos dependientes de la línea media sobre la parte de la interlínea glútea en su región posterior. Es sometida a una biopsia por Tru-cut siendo el resultado de anatomía patológica de Cordoma clásico. (Figura 4, Figura 5 y Figura 6). La paciente actualmente se halla en tratamiento con radioterapia. Caso # 2: Paciente femenina de 17 años, soltera, residente en Chaucha, de ocupación estudiante, con antecedentes de haber sufrido caída desde un caballo hace un año y cuadro clínico de 7 meses de evolución caracterizado por dolor a nivel de la región sacra y glútea del lado izquierdo y la presencia de tumoración dura, a este nivel, la misma que aumenta de tamaño y que provoca dolor, de intensidad progresiva, añadiéndose a su sintomatología parestesias de miembro inferior izquierdo y disminución de la fuerza muscular. El examen físico evidenció presencia de tumoración dura, fija de unos 14 cm en región sacra y glúteo izquierdo, dolorosa a la palpación , con disminución de la fuerza y sensibilidad de miembro inferior izquierdo. La TAC muestra lesión ocupativa de aspecto lítico y que compromete casi en su totalidad de el sacro en la porción media inferior izquierda con compromiso también de la articulación sacroilíaca y parte del ilíaco izquierdo, con compromiso importante de los tejidos blandos y continúa en sentido céfalo-caudal comprometiendo parte del coxis y el agujero ciático mayor del lado izquierdo (Figura 7, Figura 8). Es sometida a una biopsia con tru-cut obteniéndose dos milimétricos fragmentos de tejido cuyo resultado histopatológico es de cordoma clásico (Figura 9). Caso # 3: Paciente femenina de 72 años de edad, sin antecedentes de importancia, hipertensa, con cuadro clínico de 14 meses de evolución caracterizado por dolor en región lumbrosacra y coxígea de intensidad progresiva, desde hace 5 meses le provoca dificultad para la deambulación y hace dos meses paraplejía completa e incontinencia de esfínteres . En estudios de tomografía se evidencia lesiones ocupativas de aspecto tumoral infiltrante que compromete de manera difusa L4 y L5 así como también S1, produce destrucción y estallamiento del sacro con efecto de masa sobre la porción anterior sacra sobre todo al lado izquierdo (Figura 10). Se le programa para laparotomía exploradora y toma de biopsia, siendo el diagnóstico histopatológico definitivo de cordoma clásico,
(Figura 11). Debido a no tener posibilidades quirúrgicas decide abandonar el mismo. |
||
|
|
DISCUSIÓN | |
|
Los casos anteriormente mencionados reúnen todas las características clínicas y anatomopatológicas para cordoma, aunque nuestra experiencia no es grande, la centralización de neuropatología oncológica por el servicio de radiotarapia y cirugía estereotáxica, ha hecho que en poco tiempo podamos reunir tres casos que son motivo del presente análisis. Nuestros casos pertenecen al sexo femenino, con una media de edad de 58, la literatura muestra que el sexo masculino es el más frecuente, con una media de edad entre 40 y 80 años, uno de los casos es de una adolescente, situación poco frecuente. Los cordomas de la región sacra son relativamente raros, son neoplasias malignas y localmente invasivas. A pesar de la resección quirúrgica la radioterapia adyuvante y la quimioterapia, la recurrencia es común (7). En nuestros casos el dolor local y la sintomatología neurológica (paraplejia, parestesias, alteración de esfínteres) están presentes, situación debida a la localización de la neoplasia. Los estudios radiológicos muestran en todos los casos : tumor lítico e infiltración a tejidos blandos. La histopatología del tumor no muestra dificultades en la mayoría de los casos, la tipificación de los subtipos puede dar origen a la utilización de técnicas especiales tanto de histoquímica como de inmunohistoquímica, estas últimas que son de importancia para la diferenciación de la neoplasia y su origen neuroectodérmico, con la proteína S100. La utilización de citoqueratinas es útil especialmente para el diagnóstico diferencial con neoplasias de células claras como una metástasis de carcinoma renal. La experiencia del Massachusetts General Hospital incluye a 21 pacientes con cordomas de la región esfenoidal, basal y columna cervical. Seis de estos pacientes han sido seguidos durante un periodo de 3-5 años, con un único caso de recidiva local. El tratamiento fue radioterapia mediante haz de protones de precisión en el Harvard Cyclotron Laboratory. La dosis de radioterapia administrada fue del orden de 7000 a 7400 rads. Cuatro de estos pacientes fueron tratados exclusivamente mediante irradiación (8). En nuestra casuística, uno se halla en tratamiento, otro terminó su tratamiento pero con tumor residual, por lo que se halla en terapia del dolor y el tercero abandonó tratamiento. Los cordomas de localización sacral en niños, se describe que las células neoplásicas tanto en áreas convencionales de cordoma como en áreas atípicas fueron positivas para keratinas, antígeno epitelial de membrana, vimentina, proteína S100, antígeno carcinoembrionario y proteína glial fibrilar ácida. En este caso ambos pacientes fueron sometidos a resección quirúrgica y quimioterapia. Sin embargo estos mostraron un curso clínico más agresivos y fueron menos sensibles al control terapéutico. Concluyen que los cordomas sacrales en niños tienen un cuadro clínico patológico distinto denotando una alta agresividad y que deberían ser tratados como tales
(5).
|
||
|
|
PRONÓSTICO Y TRATAMIENTO | |
|
La
sobrevida es de 5-8 años, luego del diagnóstico, pueden producir metástasis
hasta un 5% de ellos, siendo los sitios mas afectados pulmones, hígado,
piel y meningeas raquídeas El
crecimiento es lento, presenta recidiva local. Debe realizarse resección
completa con amplio margen de tejido sano (cuando la localización es
sacrocoxígea). El porcentaje de recidiva es del 28% en el caso de no
haber incidido en el tumor y sube al 65% en caso de haber cortado sobre
tumor. El
estadiaje de los cordomas no ha tenido mucho valor comparado con otros
tumores óseos porque para estas neoplasias el grado es similar, las metástasis
como presentación es infrecuente y el significado pronóstico del tamaño
de la lesión es incierto (6). |
||
|
|
NOTAS AL PIE DE PÁGINA: | |
Correspondencia: Jorge Ugalde P. Departamento de Patología. Instituto del Cáncer. S.O.L.C.A. Cuenca, Ecuador. mialto:jugalde@etapa.com.ec |
||