| IV CONGRESO VIRTUAL HISPANO AMERICANO DE ANATOMÍA PATOLÓGICA |
|
CONTENIDO |
|
|
|
|
|
IMÁGENES | |||||||||||||||||||||||
|
|
|
|
INTRODUCCIÓN | |
|
La enfermedad de Kikuchi-Fujimoto, linfadenitis histiocítica necrotizante (LHN) o linfadenitis histiocítica necrotizante sin infiltración granulocítica fue descrita en 1972 de forma independiente por Kikuchi (1) y por Fujimoto y colaboradores (2) en pacientes japoneses. Posteriormente se publicaron casos en otras partes del mundo, incluido algunas series largas de casos (3-9). Es un proceso anatomoclínico que afecta sobre todo a mujeres, habitualmente en la tercera década aunque se ha descrito también en otras edades. Se caracteriza por una o varias adenopatías cervicales frecuentemente dolorosas y que suelen asociarse a fiebre (en el 30 a 50% de los casos) y leucopenia (en el 25 a 58%). Su curso es autolimitado, desapareciendo habitualmente en 2 a 3 meses y siendo raro que recidive (se describe recidiva en aproximadamente el 3% de los casos, alguna vez hasta 12 años después). Los ganglios afectos presentan una morfología peculiar que es diagnóstica pero cuya diferenciación con los procesos linfoproliferativos o el lupus eritematoso sistémico (LES) puede resultar extremadamente difícil en algunos casos (3-9). La etiología de la LHN es desconocida. Se le ha atribuido un origen autoinmune relacionándolo con el LES (6, 10-14). También se le ha atribuido un origen infeccioso, asociándolo con diversos microorganismos (Yersinia enterocolítica, Brucella, Toxoplasma), sobre todo con virus (virus de Epstein-Barr, Herpes Virus 6, Herpes Virus 8, HTLV1 y Parvovirus B-19) (3, 5, 15-23). Por tanto, no parece existir un único microorganismo con el que se le relacione específicamente. Menos frecuentemente puede haber afectación de ganglios no cervicales o poliadenopatías múltiples (se describen poliadenopatías en un 1,3 a 22,2% de los casos (3, 6, 9, 24). En raras ocasiones los pacientes pueden presentar pérdida de peso, vómitos, hepatoesplenomegalia, rash cutáneo, sudoración nocturna, dolores músculo esqueléticos, dolor torácico o abdominal o síntomas neurológicos (4, 6, 24, 25). La rara afectación extranodal está documentada en la piel, miocardio y médula ósea (4, 22, 24-29). Las manifestaciones cutáneas se ha descrito en un 2 a 40% de los pacientes pero son pocos los casos publicados con documentación histológica (22, 25). Se describen como máculas, pápulas o placas, únicas o múltiples, en la cara, cuero cabelludo, tronco o extremidades superiores, que se resuelven paralelamente a las adenopatías (25). Solo en muy raras ocasiones la LHN tiene un curso fatal (29-31). |
||
|
|
MATERIAL Y MÉTODOS | |
|
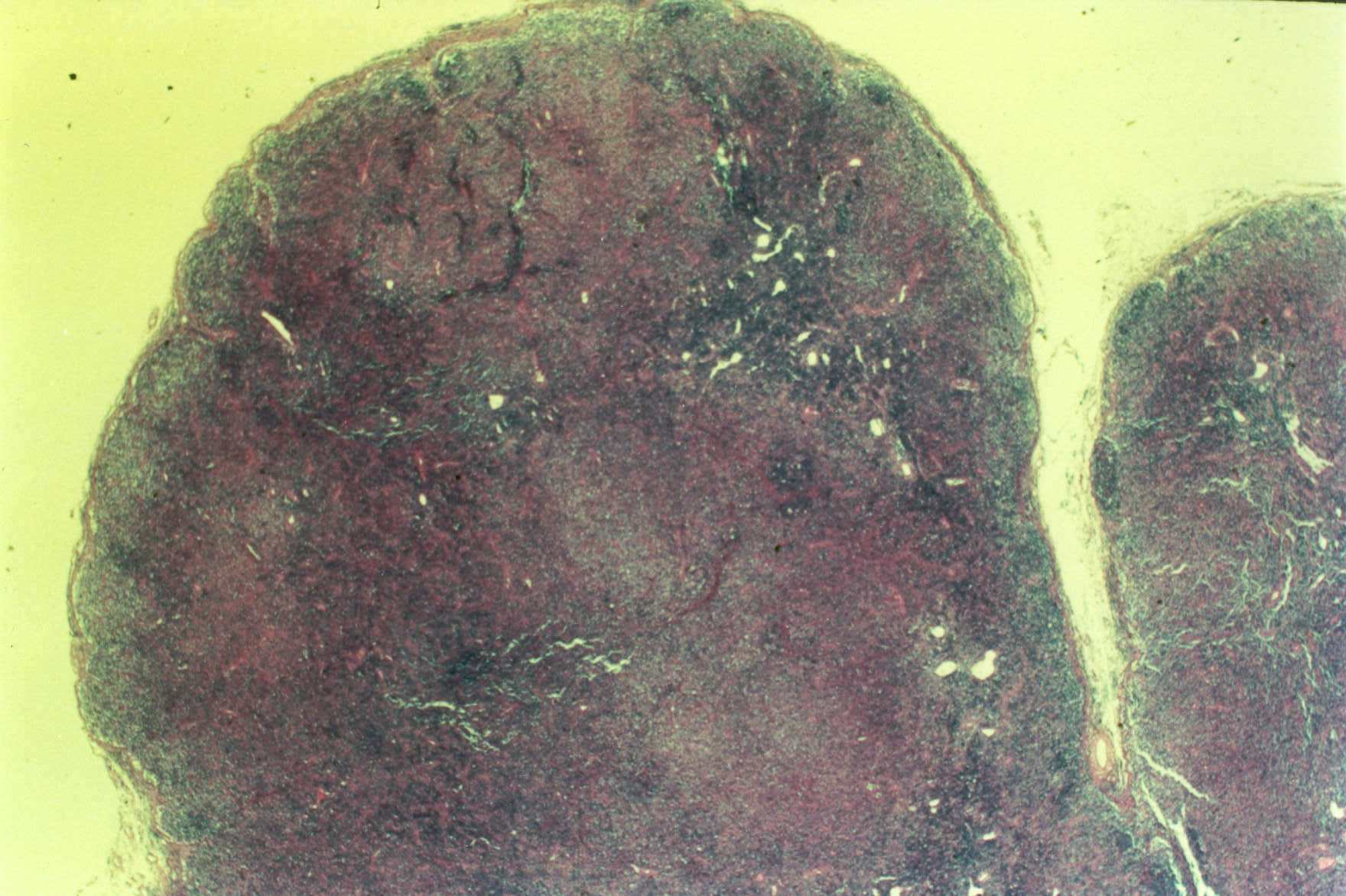
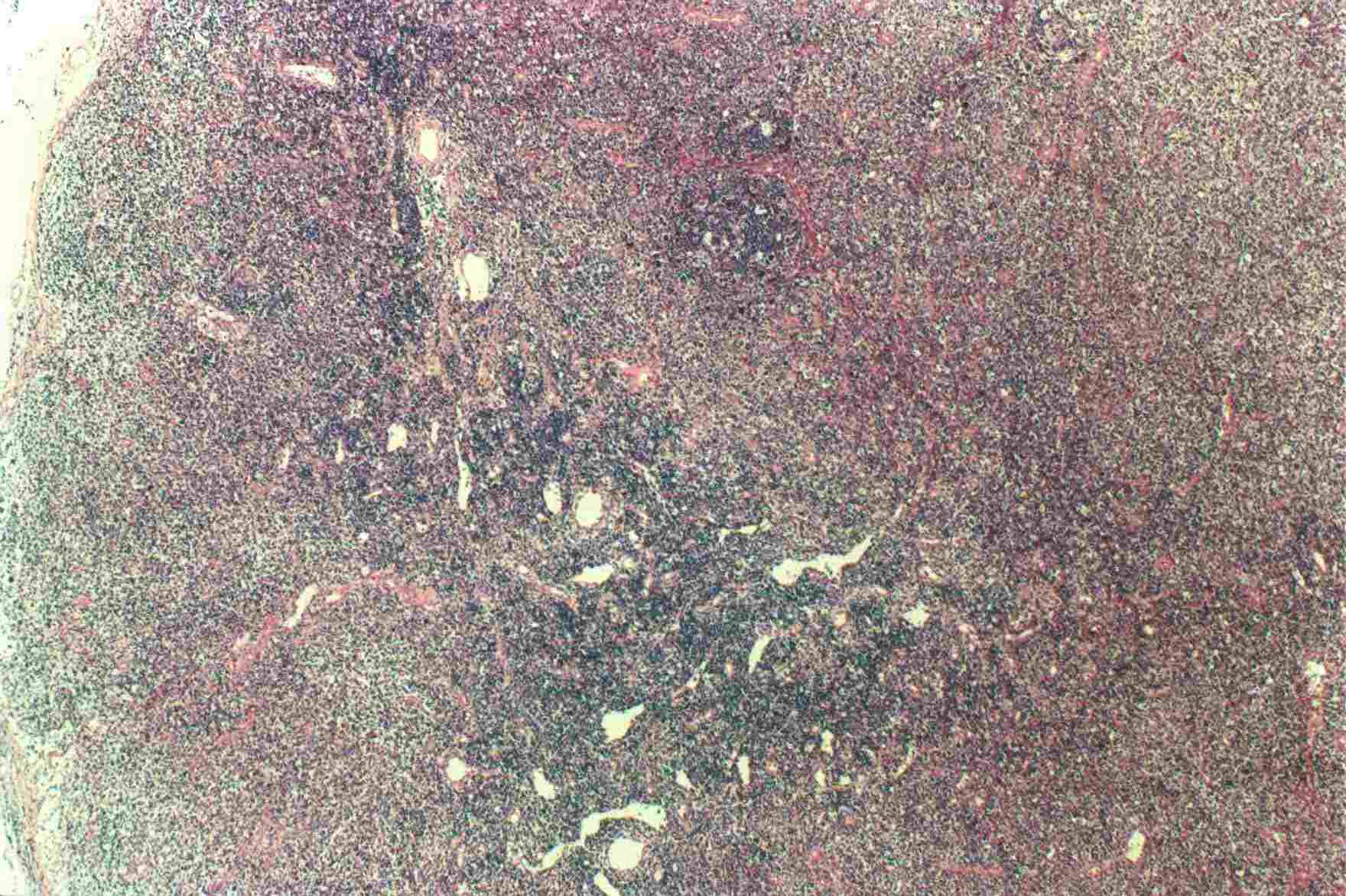
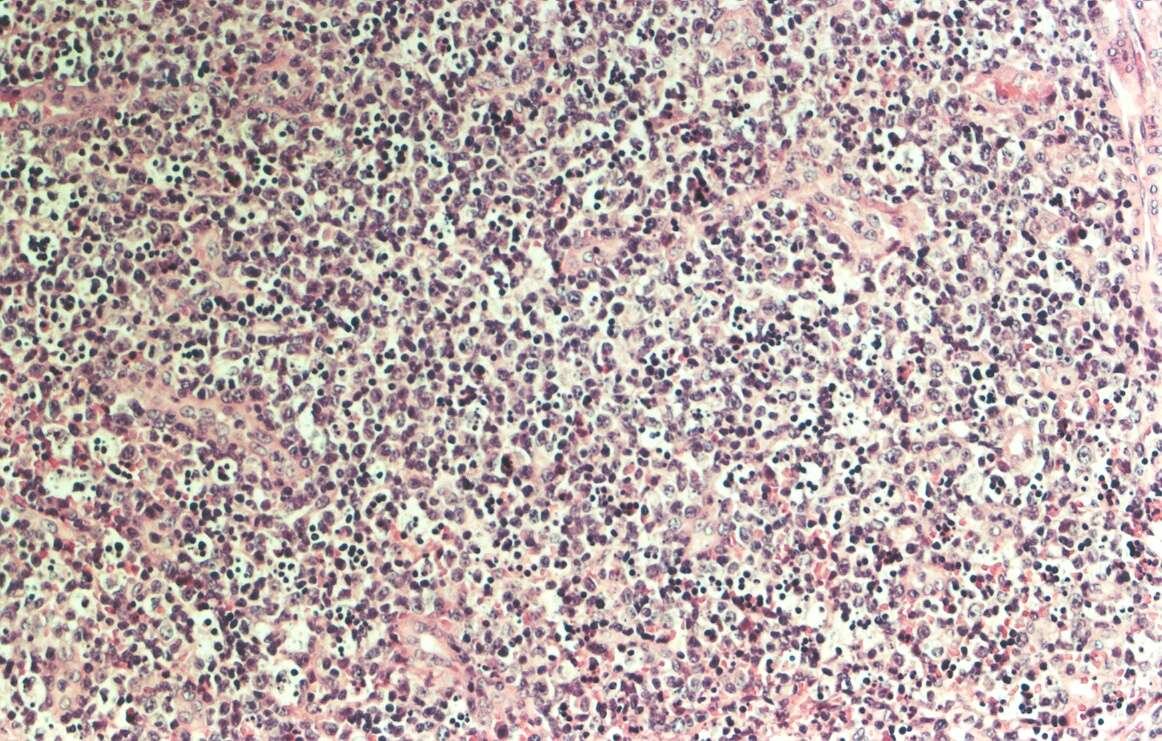
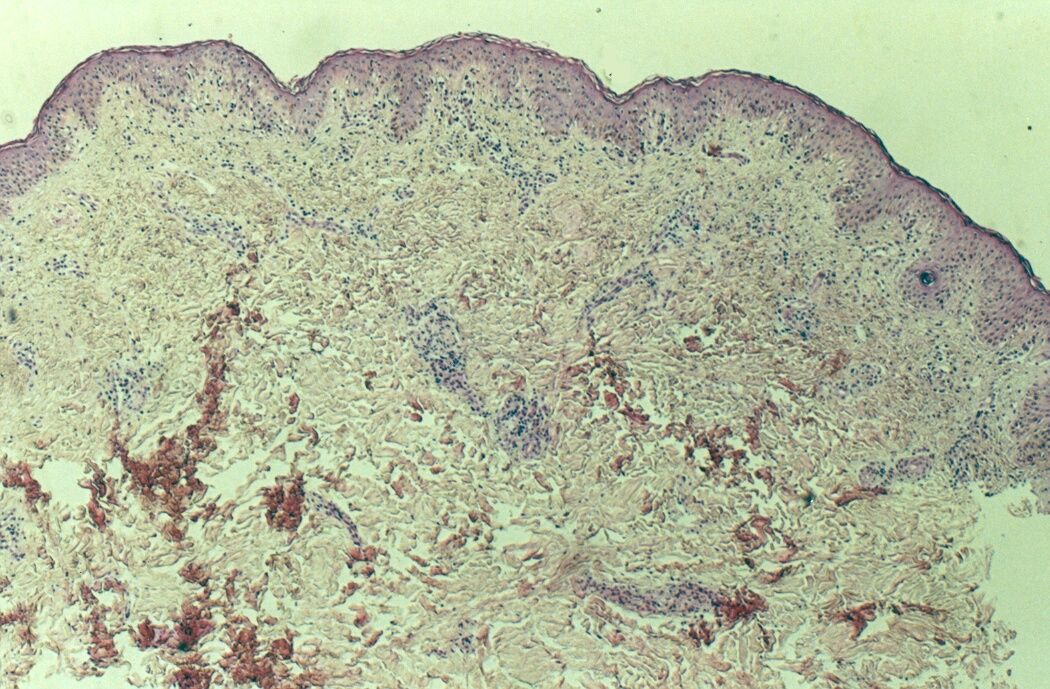
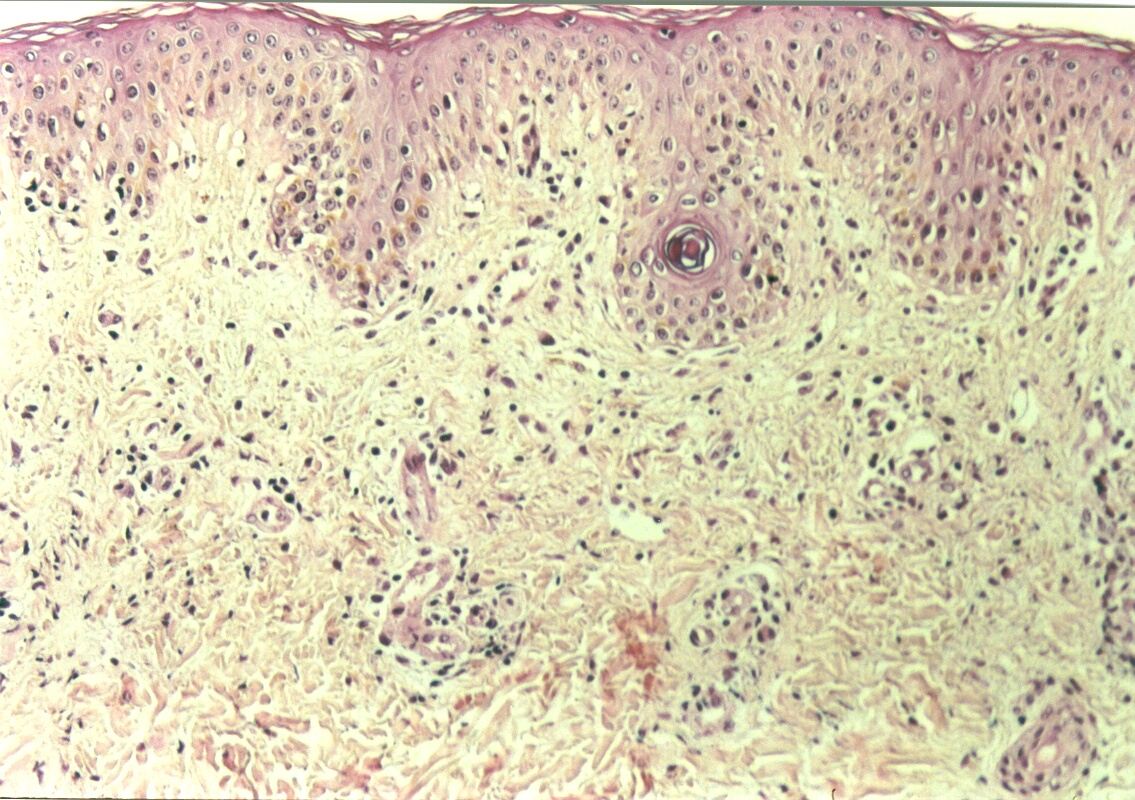
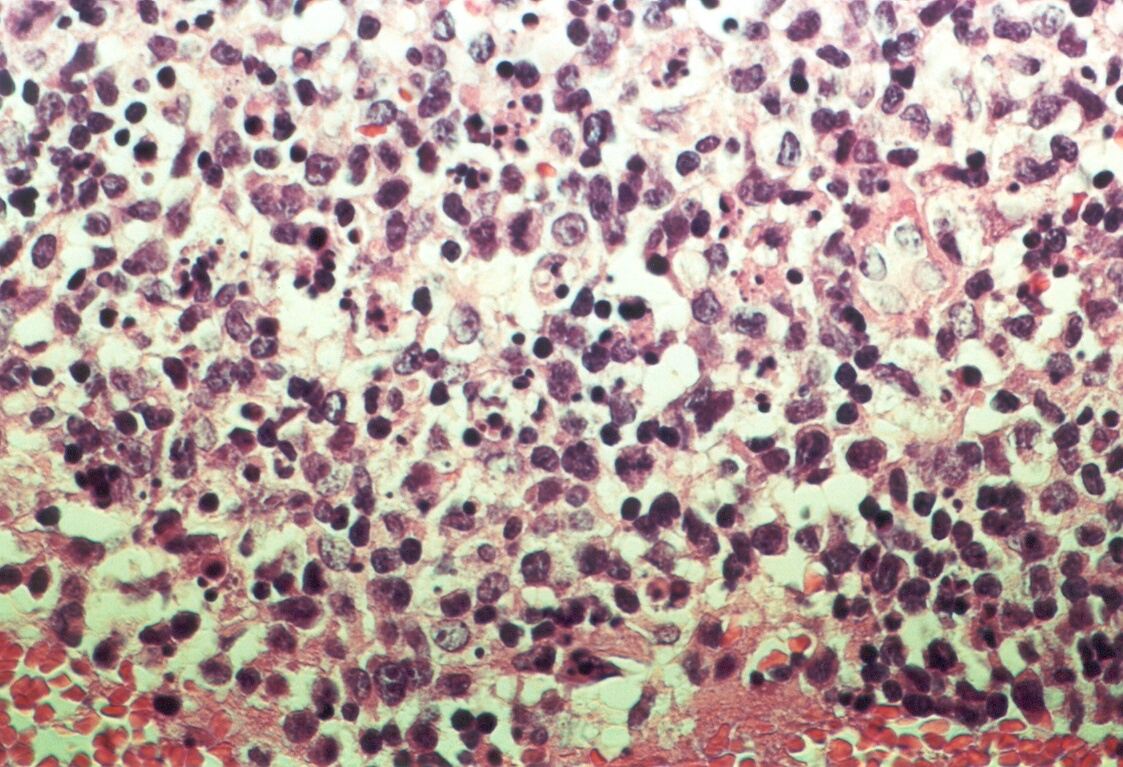
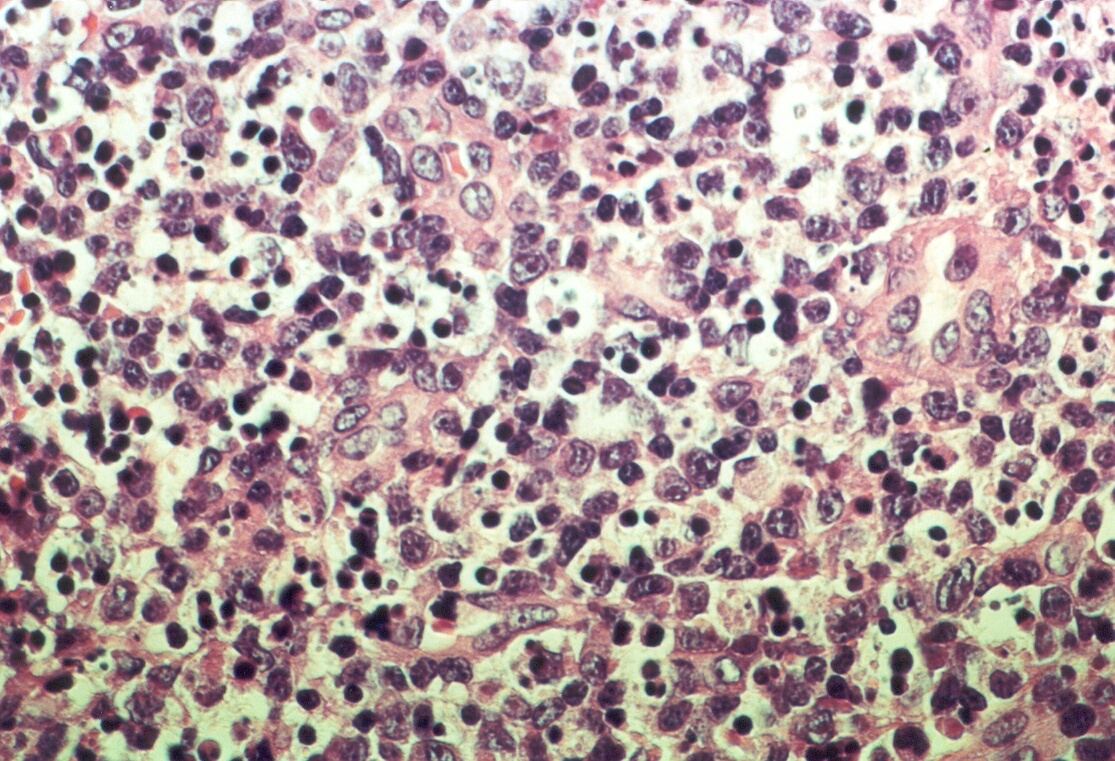
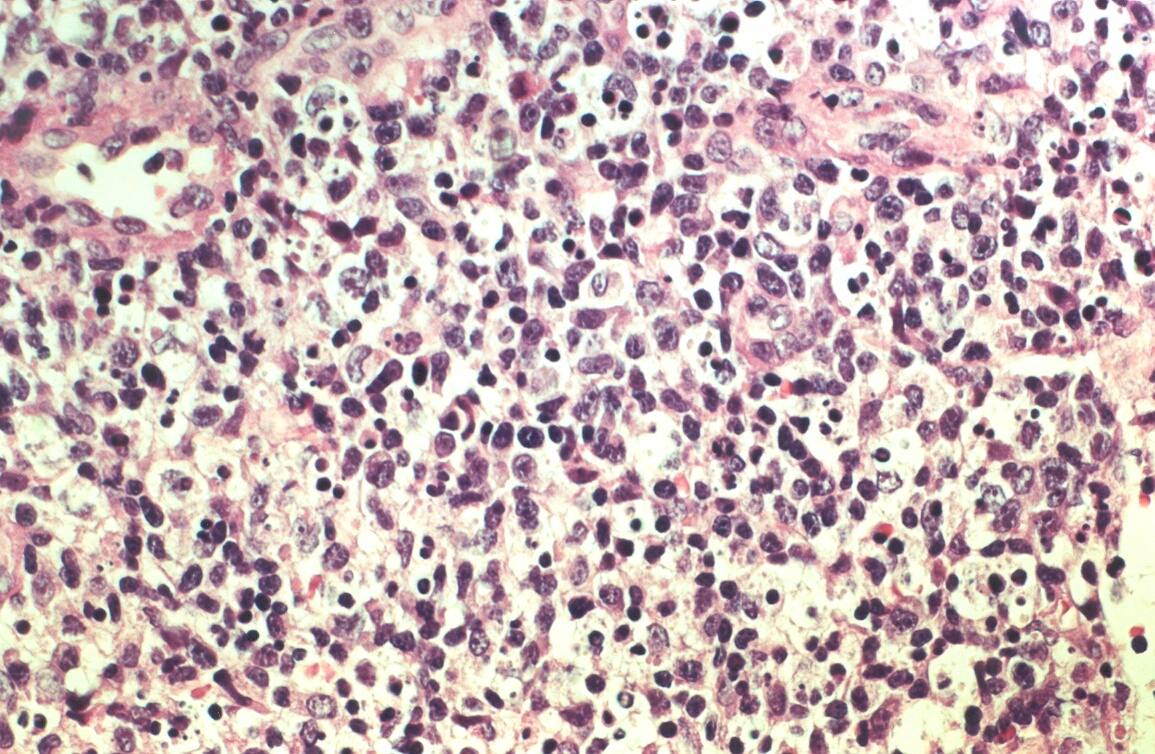
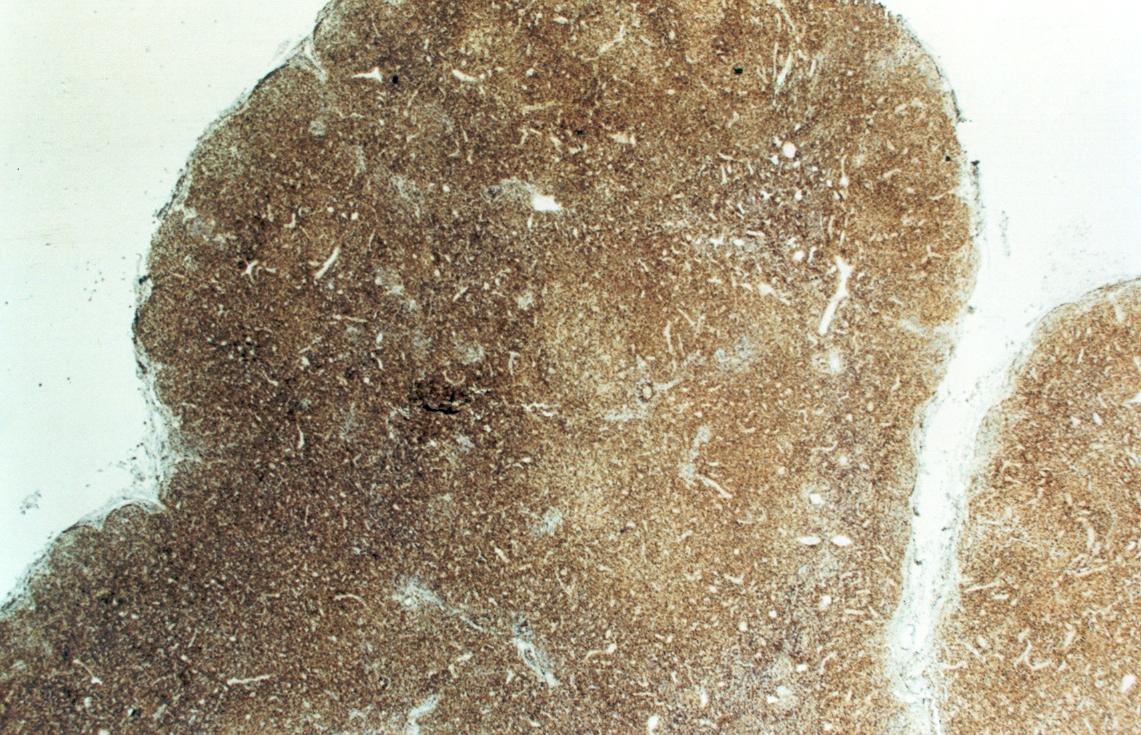
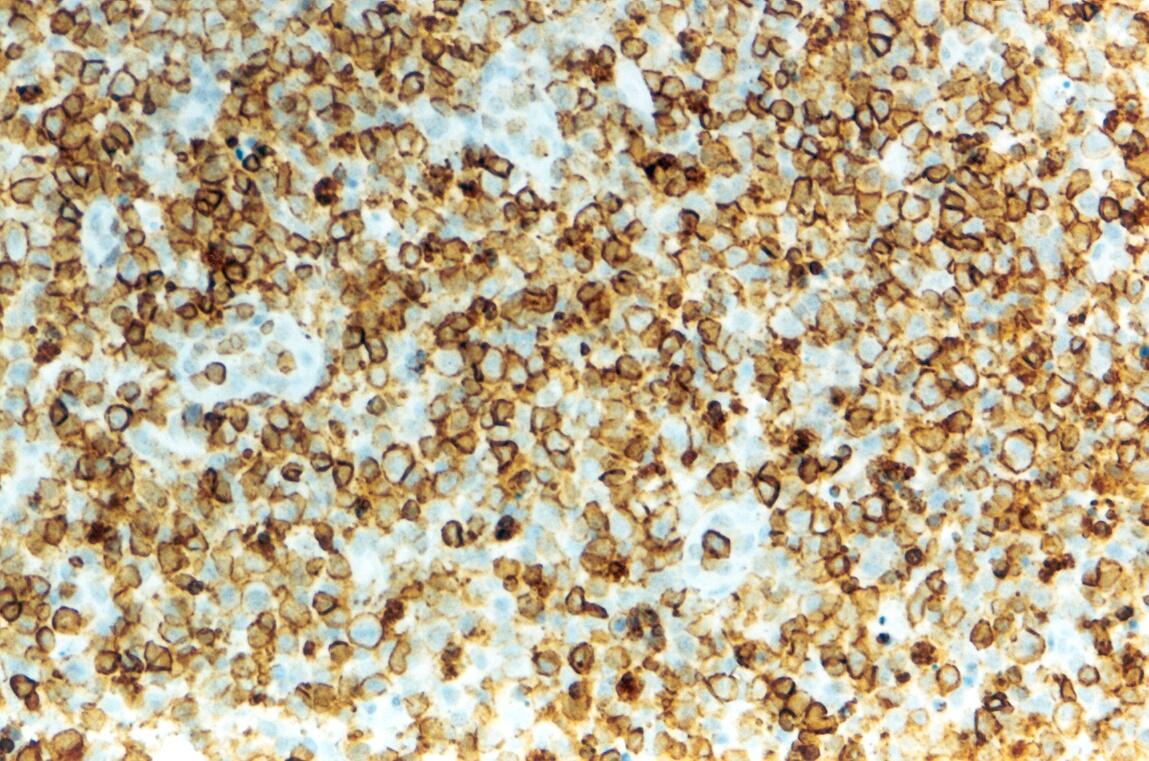
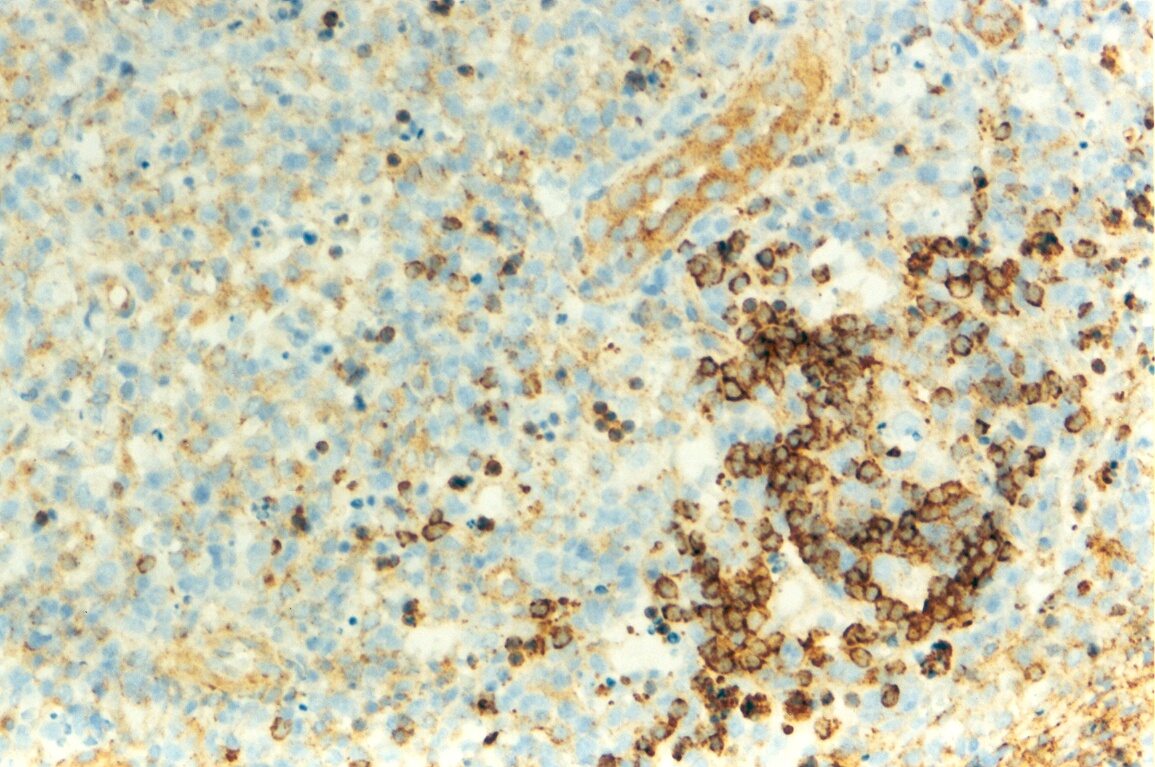
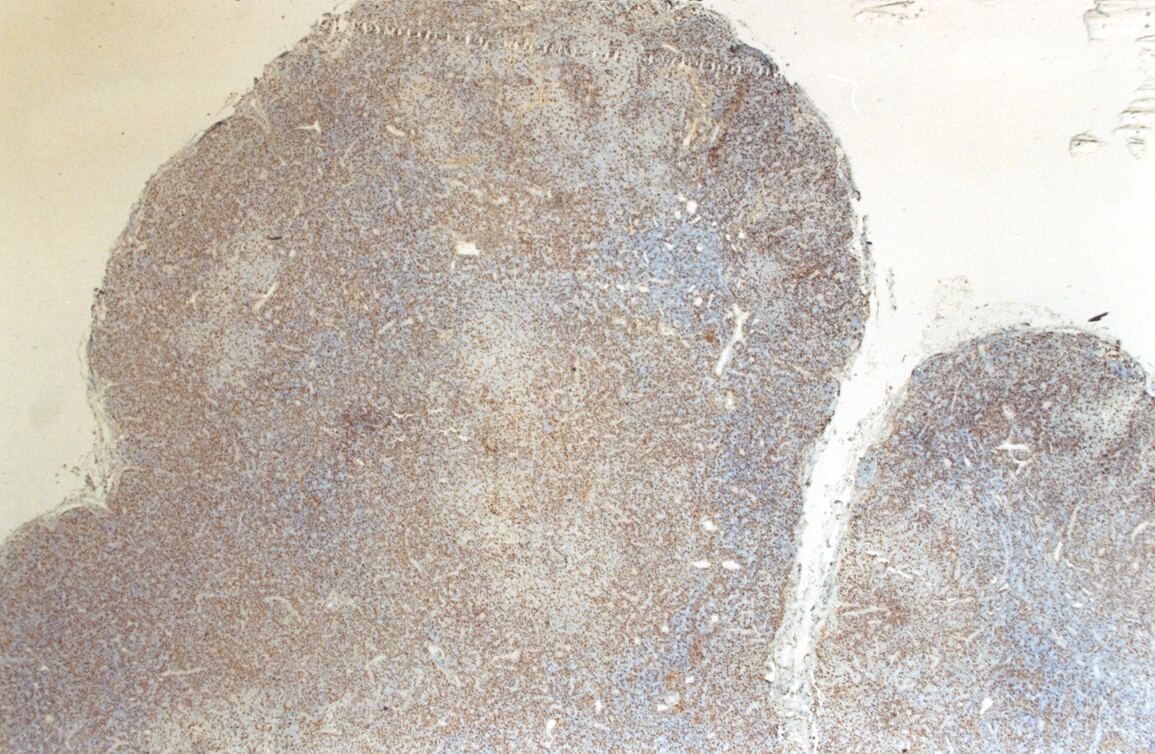
Mujer de 15 años que ingresa por cuadro de fiebre de hasta 40ºC de 11 días de evolución, síndrome constitucional, tos no productiva y exantema maculoso en tórax y abdomen. En ecografía de urgencia se observó hepatoesplenomegalia y adenopatías (subesplénicas, hiliares hepáticas y en tronco celiaco). La paciente no presenta hábitos tóxicos, alergias conocidas ni otros antecedentes personales de interés. Al ingreso se objetiva hepatomegalia palpable (a 3 cm) y poliadenopatías dolorosas a la palpación (de hasta 3 cm) en regiones laterocervical, preauricular, axilar e inguinal. El hemograma, la bioquímica general, la radiografía de tórax y los hemocultivos no muestran alteraciones relevantes. El día 2 de su ingreso la paciente desarrolla disnea e insuficiencia respiratoria gasométrica grave en relación con la aparición en la radiografía de tórax de unos infiltrados pulmonares intersticiales que llegan a ser panlobulares en 24 horas, siendo necesario el traslado a UCI e instaurándose ventilación mecánica. En su estancia en UCI desarrolla anemia (normocítica-normocroma) y plaquetopenia importantes, elevación de enzimas hepáticas, alteración de la coagulación y compromiso hemodinámico grave. En un TAC se observan adenopatías mediastínicas y retroperitonerales (de hasta 2 cm) además del infiltrado intersticio-alveolar pulmonar bilateral. Se realiza biopsia de ganglio linfático submaxilar y se instaura tratamiento empírico con corticoides a dosis altas y antibioterapia, mejorando la paciente clínica y analíticamente de forma rápida. Tras 9 días de ventilación mecánica ésta se retira y el día 17 de su ingreso hospitalario regresa a planta. El día 21 desarrolla un nuevo episodio febril con adenopatías, exantema cutáneo torácico y abdominal y malestar general, que desaparece a las 48 horas tras incrementar la dosis de corticoides. Se realiza biopsia cutánea. El día 34 es dada de alta asintomática pero regresa el día 36 por nuevo episodio febril con adenopatías y malestar general que desaparece de nuevo a las 48 horas tras incrementar la dosis de corticoides. Es dada de alta asintomática el día 61 tras su primer ingreso. A lo largo de su estancia hospitalaria los siguientes estudios fueron negativos: hemocultivos, serología y carga viral a VIH, serologías a virus de Epstein-Barr, Citomegalovirus, Adenovirus, Herpes simple, Varicela-Zoster, Rickettsias conorii y typhi, Coxiella, R. Henselae, Mycoplasma, Legionella, Yersinia, Toxoplasma y Criptococo, determinaciones de ANA y ANCA, punción lumbar (bioquímica, citología y microbiología), estudio de sangre periférica para parásitos, ecocardiografía y fibrobroncoscopia. El ganglio linfático medía 2,2 cm. Presentaba una cápsula ganglionar intacta y extensas áreas sobre todo paracorticales pero también corticales y medulares difusamente borradas, de forma que a bajo aumento se observaban unas áreas ganglionares más pálidas que otras (Figura 1 y Figura 2). A mayor aumento se observó que la población celular era polimorfa y estaba constituida por abundantes linfocitos e histiocitos, en asociación a extensa cariorrexis (Figura 3). No se observaron focos de necrosis coagulativa pese a estudiar numerosos cortes seriados del ganglio Tampoco existía un componente polimorfonuclear relevante de ningún tipo, ni presencia relevante de células plasmáticas. Los linfocitos eran muy abundantes y mayoritariamente grandes y de aspecto inmunoblástico (con escaso citoplasma y con núcleo grande con membrana nuclear irregular y uno o varios nucleolos grandes). Los histiocitos mostraban frecuentemente núcleo en forma de semiluna y tenían abundante citoplasma que comúnmente contenía restos celulares por la fagocitosis de gran parte de la cariorrexis. También se observaban otros macrófagos con cuerpos tingibles y algunos monocitos plasmocitoides (estos últimos eran células de tamaño mediano a grande, con núcleo redondeado vesicular con cromatina fina y nucleolo pequeño o ausente y con un pequeño ribete excéntrico de citoplasma sin zona de Golgi pálida). Las mitosis eran abundantes (Figura 4, Figura 5 y Figura 6). El estudio inmunohistoquímico contribuyó de forma esencial a la correcta identificación de las distintas poblaciones celulares. Reveló que la mayoría de los linfocitos eran T (positivos para CD3 y CD45RO) y señaló su enorme abundancia por todo el ganglio (Figura 7 y Figura 8), mientras que los linfocitos B (positivos para CD20 y CD79a) eran bastante más escasos y se disponían sobre todo en los restos corticales (Figura 9 y Figura 10). Los histiocitos - incluyendo a los monocitos plasmocitoides - expresaban CD68 (KP1) (Figura 11 y Figura 12). La biopsia cutánea mostró en la dermis leve edema papilar y discretos infiltrados linfohistiocitarios perivasculares superficiales y profundos, y en la epidermis escasos queratinocitos necróticos aislados, focos de espongiosis y exocitosis y moderada degeneración vacuolar de la capa basal. Se identificaron algunos fragmentos nucleares en epidermis y dermis superficial. La mayoría de las células de los infiltrados fueron positivas para CD68 (KP1) (Figura 13, Figura 14 y Figura 15). |
||
|
|
DISCUSIÓN | |
|
La LHN presenta un amplio espectro morfológico que todavía en la actualidad es confundido frecuentemente con otros procesos (32). Las poblaciones celulares que lo componen se dividen en tres grupos:
|
||
|
|
NOTAS AL PIE DE PÁGINA: | |
Correspondencia: Víctor M. Castellano Megías. Hospital Universitario de Valme. Servicio de Anatomía Patológica. Sevilla, España. mailto:apavcm@valme.sas.junta-andalucia.es |
||