| IV CONGRESO VIRTUAL HISPANO AMERICANO DE ANATOMÍA PATOLÓGICA |
|
CONTENIDO |
|
|
|
|
|
IMÁGENES | |||||||||||||||
|
|
|
|
INTRODUCCIÓN | |
|
Las fibromatosis representan un grupo de lesiones proliferativas no neoplásicas que comprende un 12% de los tumores de partes blandas identificados en la población pediátrica (1). El término fibromatosis generalizada congénita fue introducido por Stout en 1954 (2). En 1981 Chung y Enzinger (3) emplearon por vez primera la denominación miofibromatosis infantil, describiendo las características clínico patológicas del cuadro. La revisión más amplia de casos publicados corresponde a Wiswell (170) (4). |
||
|
|
MATERIAL Y MÉTODOS | |
|
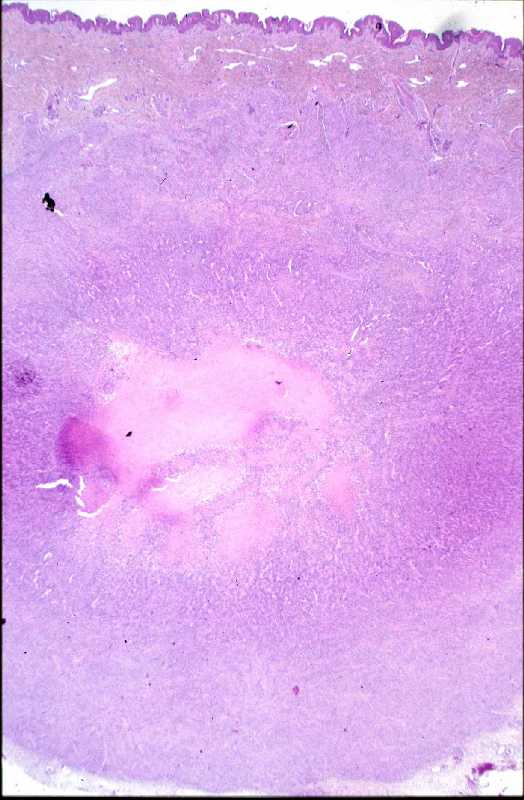
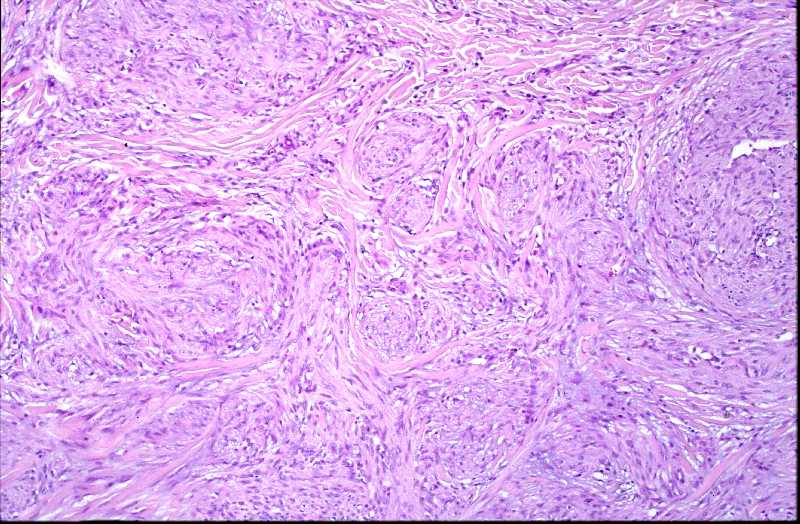
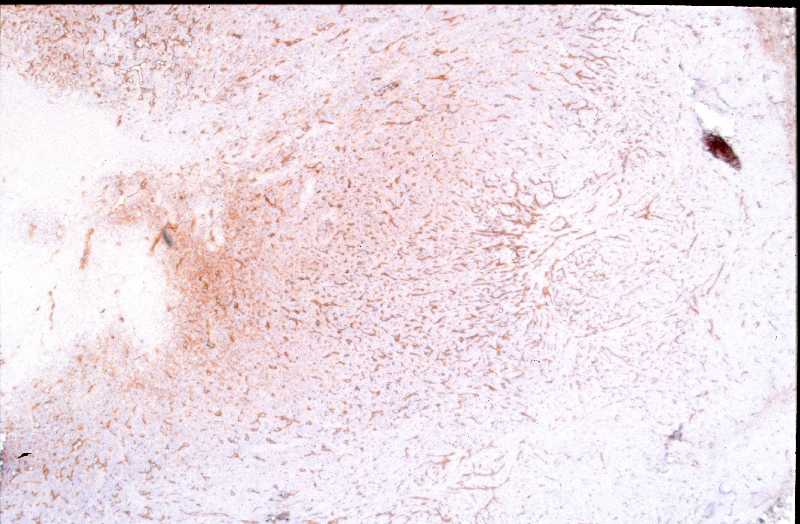
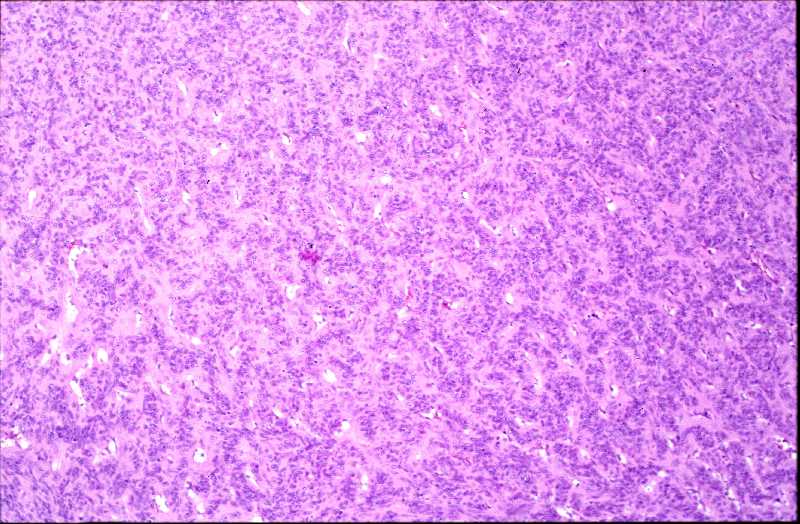
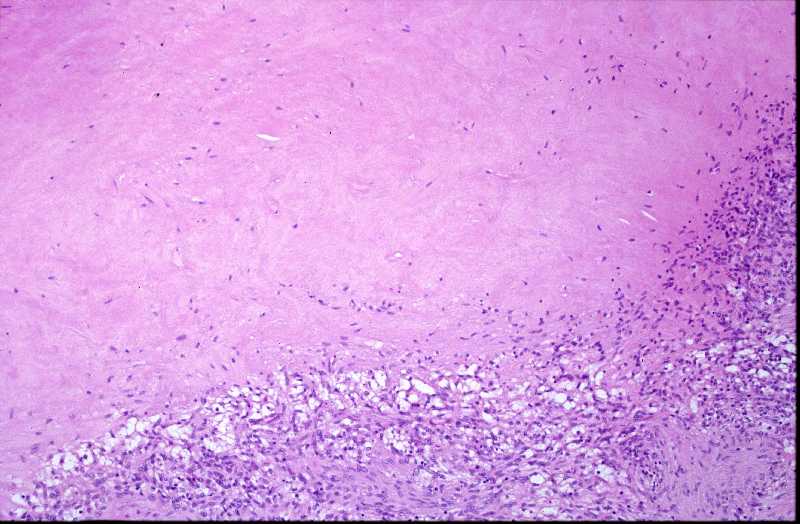
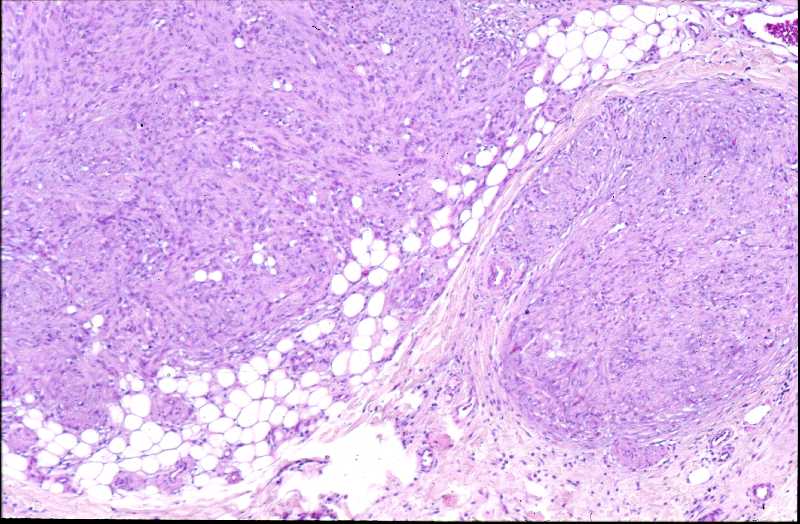
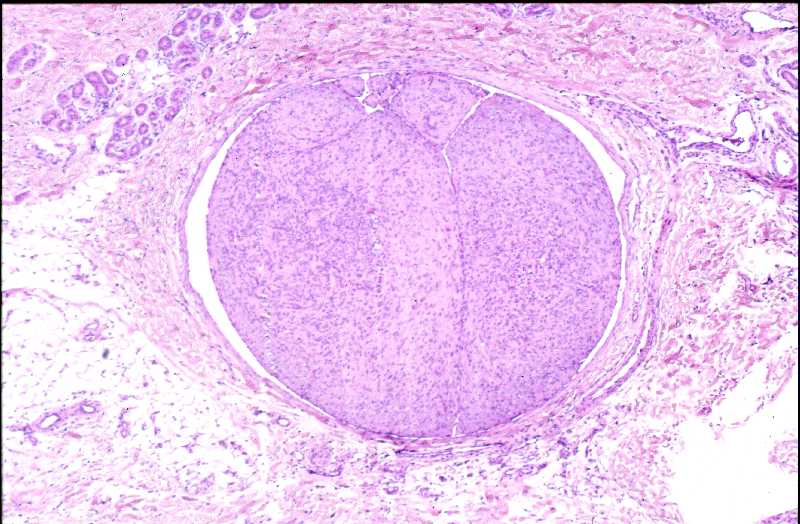
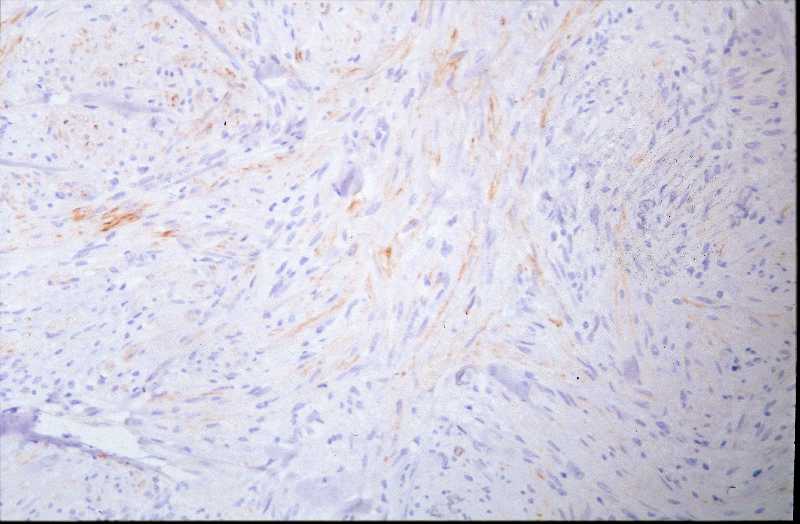
Niña de 9 meses de edad que consulta por presentar, desde 5 meses antes, una lesión nodular violácea, dura e infiltrada, pero desplazable y no adherida a planos profundos, situada en cara posterior de hombro izquierdo (Figura 1), que ha aumentado progresivamente de tamaño hasta dos meses antes, en que se ha estabilizado. Se realiza exéresis de la misma, siendo remitida con el diagnóstico clínico de dermatofibroma versus pilomatrixoma. La pieza remitida correspondía a una elipse de piel de 1.3x1x1 cm., sin lesiones relevantes en la superficie epidémica. Al corte se observa una lesión nodular que ocupa dermis e hipodermis, de coloración blanquecina y consistencia firme, de 1 cm. de diámetro máximo. La tumoración alcanza el margen profundo de resección. El estudio histopatológico mostró, a nivel de dermis y tejido celular subcutáneo, una lesión tumoral relativamente circunscrita, aunque de límites imprecisos, en la que se aprecia una arquitectura “zonal “ (Figura 2). En la periferia es posible observar una proliferación de células fusiformes, dispuestas en fascículos entrelazados o arremolinados, de patrón “leiomiomatoso” (Figura 3); en la zona intermedia se aprecian células de pequeño tamaño, separadas por espacios vasculares irregulares, de patrón “hemangiopericitoide” (Figura 4); finalmente, existe una zona central de hialinización (Figura 5). No existía atipia celular, siendo infrecuentes las mitosis. En la periferia, la tumoración muestra tendencia a infiltrar el tejido adiposo (Figura 6) y los fascículos musculares presentes en el borde profundo (Figura 7), que se encuentra afecto. Asimismo, se aprecian imágenes de crecimiento intravascular (Figura 8). Se realizaron técnicas de inmnohistoquímica , apreciándose expresión en las células proliferantes de Vimentina, Actína, (Figura 9), muy escasa de p53, reactividad únicamente en los vasos para CD34 (Figura 10) y negatividad para citoqueratinas (AE1/AE3), desmina y S-100. |
||
|
|
DISCUSIÓN | |
|
La fibromatosis infantil, descrita por vez primera por Stout en 1954 (2), y caracterizada por Chung y Enzinger (1981) (3) y Fletcher (1987) (5) y ampliamente revisada por Wiswell (1988) (4) es una proliferación benigna, probablemente hamartomatosa (6) de miofibroblastos, células que muestran miofilamentos intracitoplasmáticos en el estudio ultraestructural (3, 4). Fletcher sugirió que la histogenesis de la lesión correspondía al tejido muscular liso y propuso el término de leiomioblastoma (5). A pesar de que es más común entre el nacimiento y los dos años de edad, constituyendo el tumor fibroso más frecuente en la infancia (4), puede aparecer en la edad prepuberal (7) e incluso existen casos descritos en adultos (8, 9, 10, 11). Se han descrito casos familiares, generalmente con herencia autosómica dominante (12). La formas solitarias aparecen con mayor frecuencia en niñas, mientras que la formas múltiples suelen mostrar preferencia por el sexo masculino (3). Las localizaciones más frecuentes se sitúan en cabeza y cuello, y la afectación dérmica o subcutánea es la más común (13). Pueden aparecer también en situación intramuscular o intraósea (18), e incluso visceral (14). Suele tratarse de lesiones nodulares únicas, pese a que en las primeras descripciones se destacó más la afectación múltiple (15), y en este último caso frecuentemente están limitadas a tejidos blandos y huesos (16). A nivel visceral los órganos más comúnmente afectos corresponden al corazón, pulmones y sistema gastrointestinal (1, 4). El tamaño varía entre 0,5 y 10 cm. (15, 16), y pueden recordar a hemangiomas, cuando son superficiales, por su abundante vascularización (16). El cuadro histológico se caracteriza por un patrón bifásico, constatándose áreas fusocelulares periféricas y zonas internas con patrón hemangiopericitoide, que pueden estar centradas por áreas de hialinización. No son infrecuentes tampoco los focos de necrosis, calcificación e incluso, osificación (4). El tejido celular subcutáneo y el músculo esquelético pueden estar infiltrados por la proliferación celular (16). Los elementos que proliferan expresan vimentina, actina y proteína S-100, en tanto que son negativos para desmina (15, 17). Esta entidad se hace subsidiaria de diagnóstico diferencial con dermatofibromas, leiomiomas y hemangiopericitomas (17). Es también preciso establecer delimitación diagnóstica con procesos más infrecuentes como el sarcoma miofibroblástico, descrito recientemente (19). Éste último tiene un curso agresivo, en contraste con el comportamiento de la miofibromatosis, que, si no existe afectación visceral, es benigno y autolimitado, pudiendo incluso regresar espontáneamente (16). |
||
|
|
NOTAS AL PIE DE PÁGINA: | |
Correspondencia: Dr. F Pinedo. Unidad de Anatomía Patológica. Fundación Hospital Alcorcón. C/ Budapest, 1. 28922 Alcorcón, Madrid, España. mailto:pinedo@fhalcorcon.es. |
||