| IV CONGRESO VIRTUAL HISPANO AMERICANO DE ANATOMÍA PATOLÓGICA |
|
CONTENIDO |
|
|
|
|
|
IMÁGENES | |||||||||||||
|
|
|
|
RESULTADOS | |
|
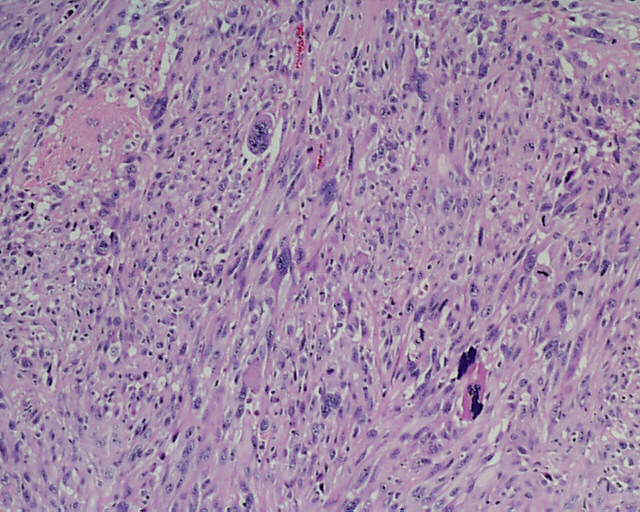
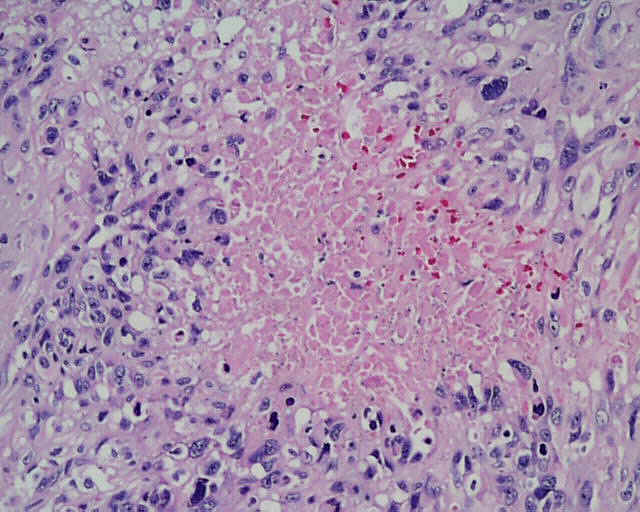
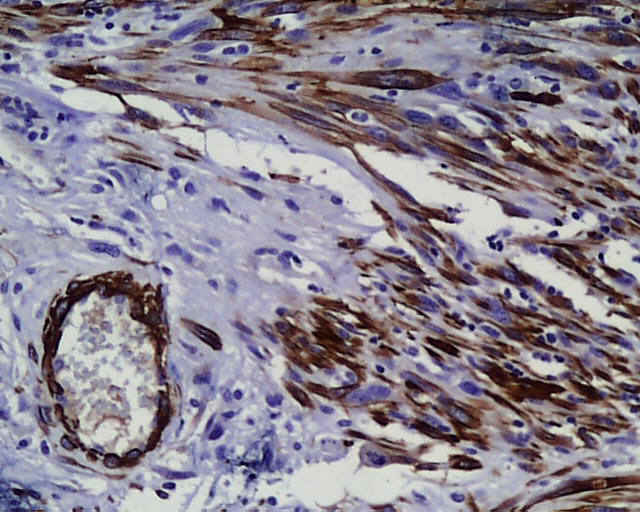
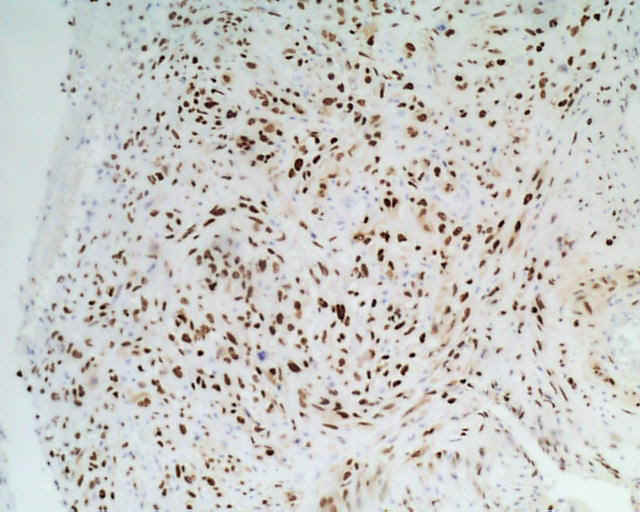
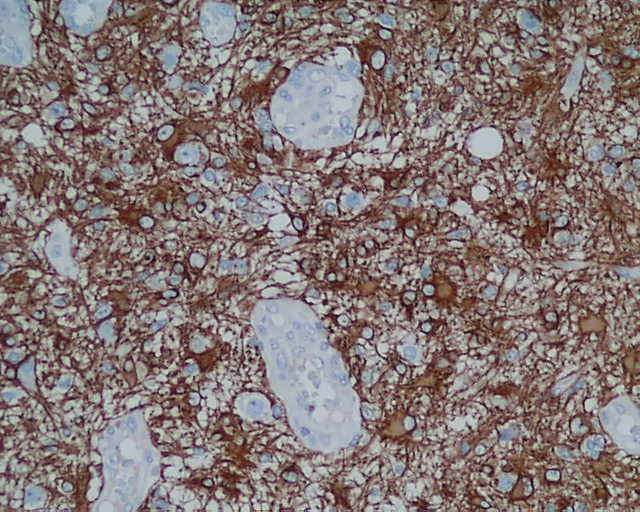
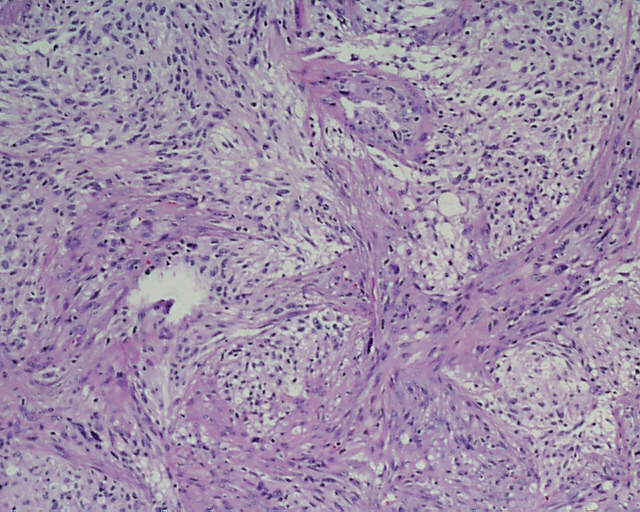
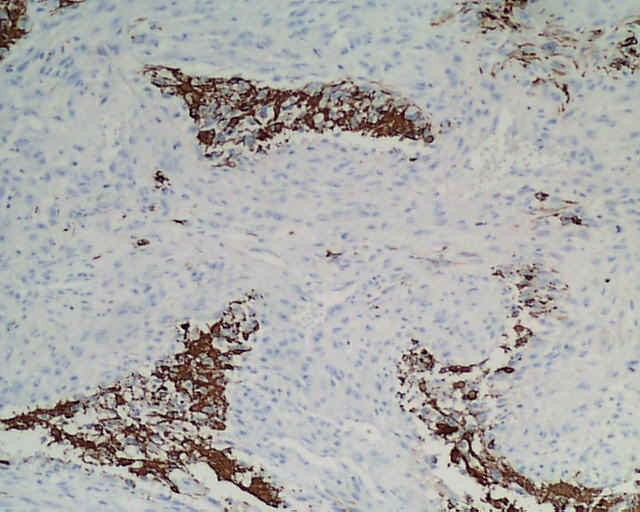
DESCRIPCIÓN MACROSCÓPICA.- Múltiples fragmentos irregulares de tejido sólido-blanquecino, con áreas hemorrágicas, el mayor de ellos de 4,5x3,5x2,5 cm. Al corte se observan áreas de apariencia mixoide, junto con áreas claramente necróticas con una consistencia mayor que los tumores gliales malignos habituales. DESCRIPCIÓN MICROSCÓPICA.- Infiltración del parénquima cerebral y meninges por proceso tumoral maligno caracterizado por una doble población celular, una de disposición fascicular y predominio perivascular con invasión de meninges, constituido por células grandes fusiformes de núcleo grande hipercromático muy pleomórfico con uno o varios nucleolos (Figura 1). Presencia de células multinucleadas gigantes atípicas, abundante necrosis e índice mitósico elevado (>50/5 CGA) (Figura 2). El patrón reticulínico muestra una disposición pericelular abundante. El estudio IHQ revela positividad para vimentina, actina (Figura 3) y CD 34 (Figura 4) y CD 68 focal, siendo negativo para la PAGF, CD31, S-100 y Desmina. Tanto la p-53 (Figura 5) como el Ki-67 se expresa en un porcentaje muy alto de células. Bien delimitado del anterior y en íntima relación al componente glial reactivo, se aprecia una proliferación glial maligna (PAGF positiva y actina negativa) con atipia nuclear, mitosis y proliferación vascular glomeruloide (Figura 6 y Figura 7). Presencia de ocasionales grupos celulares gliales tumorales aislados entre las áreas sarcomatoides del tumor (Figura 8 y Figura 9). |
||
|
|
NOTAS AL PIE DE PÁGINA: | |
Correspondencia: César Luis Ramírez Tortosa. Servicio de Anatomía Patológica, Hospital Universitario "Ciudad de Jaén". Jaén, España. mailto:cramirez@jaencolmed.org |
||