| |
INTRODUCCIÓN: La pérdida de heterocigosidad (LOH) en el cromosoma 10 es la lesión genética más frecuente en glioblastomas (GBMs), presentándose aproximadamente en un 80% de los casos. La mayor parte de los glioblastomas han perdido una copia entera del cromosoma 10, o bien muestran deleciones a nivel de 10q23-24 en un 80% de los casos, y 10q25-qter, en aproximadamente un 90% de los casos. Dos genes de particular interés de este cromosoma son PTEN en 10q23.3, y DMBT1 en 10q25.3-q26.1. Ambos son genes supresores de tumores recientemente clonados. Se detecta también LOH en astrocitomas en el locus 9p21, en un 30-50% de los astrocitomas primarios. El gen p16/MTS1/CDKN2, localizado en 9p21 es el gen candidato a estudio, y también se comporta como gen supresor de tumores.
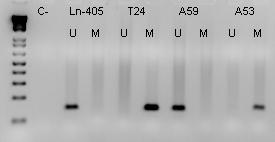
MATERIAL Y MÉTODO: Se han estudiado un total de 50 astrocitomas primarios de adultos: 22 glioblastomas (grado IV), 12 astrocitomas anaplásicos (grado III), y 16 astrocitomas de bajo grado (grado II). Los genes supresores analizados fueron p16, PTEN y DMBT1. La extracción de DNA de los tumores se realizó a partir de muestras congeladas del Instituto de Neurología de la Universidad de Tianjin, en China. La tecnología utilizada fue PCR-SSCP y secuenciación para estudio de mutaciones de p16 y PTEN; PCR diferencial para estudio de deleciones homocigóticas de p16, PTEN y DMBT1; y MSP (PCR-metilación específica) para estudio de la metilación en el promotor de p16.
RESULTADOS:
1) GEN p16: El gen p16 se estudió en 21 GBMs, no revelando mutación alguna en sus 3 exones. No se han detectado todavía mutaciones de p16 en ninguno de los 25 astrocitomas de grado II y III analizados, aunque hay PCR-SSCPs positivos en dos casos (un astrocitoma de grado II y otro de grado III) que habrán de caracterizarse mediante secuenciación. Sin embargo, 6 GBMs de 21 (29%), y 2 astrocitomas de grado II (2/12, 17%) presentaron deleciones homocigóticas de p16. También se demostró, mediante MSP que el promotor de p16 sufría hipermetilación en 2 de los 21 GBMs (9%), así como en un astrocitoma de grado II (1/12, 8%).
2) GEN PTEN: Sólo se detectaron alteraciones de PTEN en los GBMs, en concreto: 5 mutaciones en un total de 22 GBMs (23%), así como 3 deleciones homocigóticas entre los 22 GBMs (14%).
3) GEN DMBT1: Se detectaron deleciones homocigóticas de DMBT1 en 1 de 14 GBMs, y 2 de 12 astrocitomas de grado II. El estudio se hizo extensible a 20 líneas celulares de tumores del sistema nervioso (9 astrocitomas, 7 meduloblastomas/PNET, 3 neuroblastomas, y un neuroglioma) de las cuales 3 mostraron deleciones de DMBT1: un astro-oligodendroglioma, un neuroglioma y un neuroblastoma.
CONCLUSIONES:
1) Tras estos estudios concluimos que los genes p16, PTEN y DMBT1 muestran alteraciones (mutaciones, deleciones homocigoticas, e hipermetilación de promotor en el caso de p16) que producen su propia inactivación funcional en astrocitomas de bajo y alto grado. Las frecuencias de tumores alterados corresponden a 13/20 GBMs (65%), y 5/12 astrocitomas de grado II (42%). Entre los astrocitomas anaplásicos (grado III) sólo encontramos una sospecha de mutación de p16.
2) En los GBMs, la proporción de lesión del gen PTEN y del p16 es la misma, mientras que DMBT1 se ve menos dañado (8/20 p16, 7/20 PTEN, 1/14 DMBT1, 3/20 PTEN y p16 en el mismo caso). Entre los astrocitomas de bajo grado, hay 4/12 lesiones de p16, y 2/12 del gen DMBT1 (en 1/12 la lesión es en los dos genes); PTEN no mostró alteraciones en astrocitomas II.
3) Las alteraciones de PTEN sólo se detectan en GBMs. Tal vez una lesión de PTEN detectable en un astrocitoma anaplásico nos haría pensar que ese astrocitoma puede evolucionar a GBM, y, por tanto presentar un peor pronóstico. Pero eso queda por ser demostrado.
4) p16 es el segundo gen más frecuentemente mutado en nuestra serie, y curiosamente no aparece en astrocitomas anaplásicos y sí en astrocitomas de grado II, lo cual hace pensar que los tres tipos de astrocitomas se comportan como entidades nítidamente separadas en su histogénesis, es decir como grupos de tumores primarios, no sujetos a progresión tumoral desde grados II ó III a grado IV.
5) Finalmente, DMBT1 parece tener una participación menor en la génesis de astrocitomas, por lo que, si bien el esfuerzo central de la investigación molecular sobre astrocitomas debe centrarse más en las funciones que ejercen PTEN y p16 como reguladores del ciclo celular, no debemos dejar de lado la posible participación de DMBT1 en un subgrupo de astrocitomas, lo cual podría ser condicionante de su pronóstico o especificidad del tratamiento.
Palabras clave:
astrocitomas | glioblastomas | biología molecular | PCR-metilación específica |
p16/MTS1/CDKN2 | DMBT1 | PTEN | 10q23.3
|