| IV CONGRESO VIRTUAL HISPANO AMERICANO DE ANATOMÍA PATOLÓGICA |
|
CONTENIDO |
|
|
|
|
|
IMÁGENES | ||||||||||
|
|
||||||||||
|
|
INTRODUCCIÓN | |
| El Tumor Miofibroblástico Inflamatorio (TMI), también conocido como Pseudotumor inflamatorio es un tumor compuesto de células fusiformes miofibroblásticas acompañado de numerosas células plasmáticas y/o linfocitos (Weiss, 1994). El lugar originario donde se describió fue en el pulmón, aunque posteriormente se encontró en numerosas localizaciones. Existen numerosas incógnitas acerca de su patogénesis bien como un proceso reactivo de carácter exagerado o como una verdadera neoplasia. Clínicamente se suele presentar con un cortejo acompañante en forma de fiebre, sudoración nocturna, fatiga, pérdida de peso y adenomegalias, así como otras alteraciones en las pruebas de laboratorio, esto es hipergamaglobulinemia policlonal y aumento en la velocidad de sedimentación globular sugiriendo una posible causa autoinmune o infecciosa (Arber et al, 1998). Sin embargo, a pesar de estas sospechas, sólo de forma ocasional se han encontrado microorganismos bacterianos, fúngicos o virales asociados al TMI (Matsubara et al, 1988). Por otro lado, recientemente se ha descrito un nuevo miembro de la familia de los gammaherpesvirus, llamado virus del herpes asociado al Sarcoma de Kaposi (VHSK) o Virus del Herpes humano tipo 8 (VHH8) en cerca del 95% de casos de Sarcoma de Kaposi (SK) de pacientes con o sin SIDA (Chang et al, 1994; Marchioli et al, 1996; Gómez-Román et al, 1999). Este virus es especial ya que su AND codifica para proteínas que simulan proteínas de señalización intercelular y reguladoras de ciclo celular como bcl-2, ciclina-D, factores reguladores de interferón e IL-6 (Cesarman and Knowles, 1997). Tales moléculas han sido implicadas en la patogénesis del SK, y de hecho IL-6 tiene una probada función en la proliferación de las células tumorales del SK asociado a SIDA, queratinocitos, células renales mesangiales, células musculares lisas y mioblastos (Kishimoto et al, 1992). Existe un consenso creciente acerca de que el VHH8 es el cofactor infeccioso necesario para todas las formas de SK (Lin et al, 1996), aunque debe ser dilucidado todavía el carácter transformante o permisivo del VHH8 en la tumorigénesis (Gallo, 1998). Algunas comunicaciones han sugerido la importancia de IL-6 en el desarrollo del TMI
(Coffin et al, 1998), (Kishimoto et al,
1992). Sin embargo, hasta el momento no existían comunicaciones acerca de la posible relación entre VHH8 y el TMI. Describimos la presencia de secuencias de ADN de VHH8 y la sobreexpresión de citoquinas en cinco casos de TMI pulmonar
|
||
|
|
MATERIAL Y MÉTODOS | |
Descripción de los casos: Cinco casos de pseudotumor inflamatorio pulmonar fueron seleccionados de nuestros archivos del Departamento de Anatomía Patológica del Hospital Universitario Marqués de Valdecilla en Santander (España). Los detalles epidemiológicos, de localización y de seguimiento están detallados en la Tabla 1. Todos los casos fueron revisados por dos patólogos de nuestra institución de forma ciega y sólo se aceptaron los casos que cumplían los criterios de diagnóstico establecidos por la OMS (Weiss 1996). Se revisaron las historias clínicas de todos los pacientes y se recogieron todos los datos clínicos de interés. En todos los casos se llevó a cabo un seguimiento de al menos 6 meses tras la intervención. En todos los casos, procedimos a la extracción de ADN a partir de material fijado en formol e incluido en parafina. Usamos el kit NucleoSpin C+T (Macherey-Nagel Gmbh, Düren, Alemania). El ADN de cada espécimen fue obtenido en un laboratorio aislado del lugar donde se llevó a cabo la PCR. La amplificación se repitió hasta tres veces para asegurar la reproducibilidad de los resultados. Las muestras fueron analizadas de forma ciega. Se aplicaron condiciones de laboratorio estrictas y controles apropiados para evitar la aparición de contaminación y falsos resultados positivos. Todas las muestras fueron sometidas a una amplificación del exón 8 del gen inhibidor de C1 como control interno de calidad del ADN extraído. Para la detección de ADN de VHH8 usamos un procedimiento de PCR-nested como ha sido descrito previamente (Moore 96), frente a una región de 571 bp en la reacción externa (sentido y antisentido, respectivamente) KS4 (5'-AGCACTCGCAGGGCAGTACG-3') y KS5 (5'-GACTCTTCGCTGATGAACTGG-3'). La amplificación de la región externa se llevó a cabo con las siguientes condiciones: Desnaturalización inicial: 94ºC 2 min. Posteriormente 35 ciclos de 94ºC 30 seg; 60ºC 30 seg; 72ºC 45 seg. Extensión final: 72ºC 5 minutos. Se añadió 1 microlitro del producto de PCR a una mezcla de reacción interna que se amplificó durante 25 ciclos adicionales con primers frente a la región KS330233 de 233 pb KS1 (5'-AGCCGAAAGGATTCCACCAT-3') y KS2 (5'-TCCGTGTTGTCTACGTCCAG-3'). Las amplificaciones se llevaron a cabo en un termociclador Perkin-Elmer 9600. Los productos de PCR se analizaron en gel de agarosa al 2% con bromuro de etidio. La confirmación de los resultados se llevó a cabo amplificando regiones no sobrelapadas de genes mayores de cápside como ha sido sugerido por otros autores (Moore 96). Los primers utilizados fueron: set no. 1 (externo sentido 5'-AGGCAACGTCAGATGTGAC-3', externo antisentido 5'-GAAATTACCCACGAGATCGC-3', interno sentido 5'- CATGGGAGTACATTGTCAGGACCTC-3', interno antisentido 5'-GGAATTATCTCGCAGGTTGCC-3') que generan un producto de PCR de 213 bp; set no 2 (externo sentido 5'-GGCGACATTCATCAACCTCAGG-3', externo antisentido 5'-ATATCATCCTGTGCGTTCACGAC-3', interno sentido 5'- CGCATGGAGGACCTAGTCAATAAC-3', interno antisentido 5'-GGTTGTAGTCATTCTCGTCCAGGG-3' que generan un producto de PCR de 115 bp con las mismas condiciones que las señaladas anteriormente. Los productos amplificados fueron transferidos a papel de nitrocelulosa y sometidos a hibridación con el método de Southern con una sonda de 25pb marcada con [32P]-trifosfato de deoxicitidina, representando una secuencia de pares de bases comprendida entre 1078 y 1102 (Chang et al 1994). Hibridación in situ: Para la hibridación in situ se utilizó un fragmento de ADN de 115pb del ORF72 viral obtenido de un control tisular de un sarcoma de Kaposi. La generación de la sonda se realizó a partir de una PCR con los siguientes primers: P51 (5' primer; 5'-CACCCTGAAACTCCAGGC-3') y P32(3'primer; 5'-GATCCGATCCTCACATAGCG-3') El producto de PCR fue purificado (CONCERT Rapid PCR purification System, Life Technologies, Barcelona, Spain) y analizado en electroforesis. El producto fue marcado con biotina (BioPrime DNA labelling System, Life Technologies) siguiendo las instrucciones del fabricante. La hibridación in situ se practicó sobre cortes histológicos de 5 mm de espesor. Tras proceso de desparafinación y rehidratación, se bloqueó la actividad de peroxidasa endógena con peróxido de hidrógeno al 3%. Se incubaron las secciones en una solución de Proteinasa K (10 mg/ml) a 37ºC durante 30 minutos. La sonda se diluyó a una concentración de 0.1μg/ml en una solución de 10% vol/vol 20X SSC, 10% vol/vol solución de Denhardt, 2.5% salmon testes DNA (10 mg/ml), 50% vol/vol deionized formamide, 22.5% vol/vol agua destilada con 10% wt/vol sulfato de dextrano y 5% vol/vol tRNA. Se aplicaron 50 microlitros de dicha solución a las secciones y se calentaron en placa a 95ºC. Posteriormente se incubaron a 37ºC durante 16 horas en cámara húmeda. Posteriormente se realizaron varios lavados. Para visualizar la señal se utilizó un sistema de amplificación colorimétrica no isotópica con tiramida (DAKO GenPoint, DAKO Corporation) siguiendo las recomendaciones del fabricante. Los controles positivos correspondieron a biopsias cutáneas de pacientes con Sarcoma de Kaposi. Los controles negativos a biopsias cutáneas de individuos sanos. La contratinción se eralizó con hematoxilina de Harris Inmunohistoquímica: Utilizamos los siguientes anticuerpos: IgG1 anti-recombinante IL-6 humana (Genzyme. USA. Monoclonal. Dilución 1/100) e IgG1 anti-Ciclina D1 humana (DCS-6 Dako Glostrup Denmark 1/100). Se usaron sobre tejido fijado en formol e incluido en parafina. IL-6 se incubó a 4ºC toda la noche. Se utilizó recuperación antigénica mediante olla a presión y buffer citrato durante 90 segundos para IL-6 y 0,1mM EDTA pH8.0 para la CiclinaD1. Usamos el sistema de visualización EnVisionTM + de Dako en un inmunoteñidor Techmate 500-220 (Biotek, Santa Barbara, CA)
(Vyberg and Nielsen, 1998). Diaminobenzidina fue el cromógeno utilizado
|
||
|
|
RESULTADOS | |
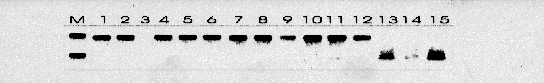
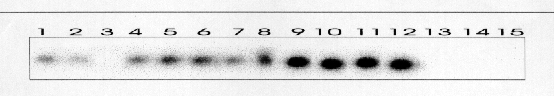
Todos los casos fueron intervenidos quirúrgicamente con el diagnóstico de pseudotumor inflamatorio. Los tumores correspondieron con grandes masas blanquecinas con apariencia al menos focalmente arremolinada y consistencia firme. Microscópicamente estaban compuestos por células fusiformes miofibroblásticas positivas para Vimentina, Actina de músculo liso y negativas para marcadores epiteliales. En tres de los casos aparecían áreas ricas en células plasmáticas y linfocitos. No se apreciaron áreas de pleomorfismo, necrosis o figuras mitóticas atípicas. El seguimiento no reveló evidencia de recidiva tumoral en ningún caso. Se detectaron las secuencias de VHH8 las tres ocasiones en las que se practicó la PCR (Figura 1). También se observó positividad en dos muestras de Sarcoma de Kaposi asociado a SIDA y Sarcoma de Kaposi pleural en un paciente con inmunosupresión. No se detectó amplificación en ninguno de los casos usados como controles negativos. La hibridación por el método de Southern confirmó estos resultados
(Figura 2). Las PCRs frente a las regiones no solapadas también resultaron positivas.
La hibridación in situ mostró positividad en núcleos de células fusiformes en los cinco casos
(Figura 3).
La inmunohistoquímica reveló positividad para IL-6 humana en la mayor parte de las células miofibroblásticas, células endoteliales y población linfoide entremezclada
(Figura 4). La CiclinaD1 humana mostró reaactividad variable en los núcleos de los miofibroblastos en los cinco casos examinados
(Figura 5).
|
||
|
|
DISCUSIÓN | |
La localización más frecuente del pseudotumor inflamatorio es el pulmón (Coffin et al, 1998). La aparición clínica puede ser de tipo insidioso o acompañarse de un cuadro con síndrome constitucional de fiebre, pérdida de peso y otras anomalías de laboratorio en un 15-30% de los casos. Estos hechos sugieren la posibilidad de que algunas citoquinas como la IL-6 pudieran estar involucradas en la patogénesis del TMI (Coffin et al, 1998). El principal componente del TMI es la célula miofibroblástica. Éstas son células ubicuas y en cualquier situación en la que ocurre un daño tisular los fibroblastos se transforman en miofibroblastos. Esta situación está bajo el control de varias citoquinas y factores de crecimiento entre los que la IL-6 es uno de los más importantes (Kishimoto et al, 1992). Por otro lado, el TMI es una fuente importante de IL-6. Este hecho puede explicar también los niveles elevados de IL-6 e IL-1β que se han hallado en sangre periférica y en tejido de un niño con un granuloma de células plasmáticas de pulmón (Rohrlich et al, 1995). Algunos autores han tratado de identificar agentes infecciosos en el seno del TMI (Matsubara et al, 1988). De todos modos, el papel de un agente infeccioso o no, desencadenante podría estar restringido a los estadios iniciales de la enfermedad. Éste podría iniciar una cascada de reacciones paracrinas en las cuales las células estromales y el entorno inflamatorio pudieran sufrir un intercambio de factores proinflamatorios y proliferantes tras los cuales el tumor llegaría a ser autónomo. La producción de factores solubles a partir de esta cascada podría inducir las manifestaciones clínicas de la enfermedad (Matsubara et al, 1988). El VHH8 se ha detectado en el 95% de los casos de Sarcoma de Kaposi (SK) de pacientes asociado o no al SIDA (Chang et al, 1994; Marchioli et al, 1996) (Gómez-Román et al, 1999). Este virus ha sido relacionado además con el linfoma de cavidades, la enfermedad linfoproliferativa benigna, hiperplasia angiolinfoide con eosinofilia, enfermedad de Castleman's multicéntrica, mieloma (Marchioli et al, 1992; Foreman et al, 1997) y sarcoidosis (DiAlberti et al, 1997). Existen asimismo casos aislados de detección de secuencias de ADN de VHH8 en pacientes sin inmunosupresión o SK (Memar et al, 1997; Trovato et al, 1999) aunque existe un acuerdo general de que los efectos patogénicos de VHH8 se potencian con la inmunosupresión. De forma llamativa VHH8 ha sido hallado en las regions interfoliculares de ganglios linfáticos hiperplásicos y en células endoteliales y neumocitos del pulmón en pacientes no inmunodeprimidos (Trovato et al, 1999). Ambas localizaciones estarían implicadas en el caso de los pseudotumores inflamatorios. Se ha comunicado recientemente la secuencia de VHH8 (Russo et al, 1996). Se ha encontrado que el virus codifica homólogos de una gran variedad e genes celulares, como Ciclina D (Cesarman et al, 1997), e IL-6 (Moore et al, 1996). La IL-6 viral se ha demostrado como funcional en ensayos de proliferación y se expresa en células infectadas por VHH8 de estirpe hematopoyética y endotelial (Moore et al, 1996). Por otro lado, la IL-6 viral puede activar el receptor para la IL-6 humana (Wan et al, 1999), y puede iniciar una cascada de reacciones paracrinas que afectan IL-6 y otras citoquinas inflamatorias implicadas en la inhibición de la apoptosis (IL-6) y la transcripción de genes implicados en la síntesis de ADN (cyclin D). Aunque en esta comunicación no hemos practicado inmunotinción para IL-6 y ciclina D viral, hemos demostrado la presencia de IL-6 y ciclina D humana mediante anticuerpos monoclonales murinos. Se ha demostrado que un anticuerpo policlonal contra IL-6 viral no reacciona de forma cruzada con la IL-6 humana (Parravicini et al, 1997) no concocemos si los anticuerpos frente a la IL-6 humana reaccionan de forma cruzada con su homólogo viral. El mecanismo oncogénico de VHH8 es desconocido hasta el momento. La mayoría de los genes reguladores de ciclo celular que son responsables de la tumorigénesis en otros virus del herpes son genes asociados a latencia
(Ganem, 1997) y cualquiera que sea el mecanismo éste podría explicarse por una reactivación viral más que por una infección primaria lítica
(Cesarman et al, 1997; Gilison and
Ambinder,1997). Como ocurre con otros virus de la familia del herpes, muchas situaciones asociadas como inmunosupresión, carga viral, coinfecciones y citoquinas proinflamatorias podrían interaccionar en la patogénesis, morfología y agresividad biológica de las enfermedades asociadas con VHH8
(Chang et al, 2000). Por otro lado, la mayoría de los homólogos de citoquinas y factores antiapoptóticos se expresan durante el ciclo lítico viral. Por tanto, dicho ciclo lítico podría contribuir inicialmente a la patogénesis tumoral
(Sun et al, 1999). Nuestros hallazgos en un tumor como el pseudotumor inflamatorio, caracterizado por un crecimiento autolimitado, con la presencia de partículas virales en muchas células tumorales y una sobreexpresión de citoquinas humanas en un entorno inflamatorio, probablemente favorecería una reactivación local de la replicación lítica de VHH8 que daría lugar a una neoplasia autónoma miofibroblástica.
|
||
|
|
NOTAS AL PIE DE PÁGINA | |
Correspondencia: José Javier Gómez-Román. Dpto. Anatomía Patológica y Servicio de Inmunología Clínica. Hospital Universitario Marqués de Valdecilla. Instituto Nacional de la Salud. Santander, España. mailto:apagrj@humv.es |
||