Palabras clave.
Miofibromatosis, congénita, fibromatosis, pediatria
Introducción.
En el período neonatal la miofibromatosis
congénita es un proceso de evolución generalmente favorable que
plantea el diagnóstico diferencial con otras neoformaciones mesenquimales
y cuyas manifestaciones clínicas dependen de la localización,
profunda o superficial, así como de los órganos afectados. Presentamos
un caso de afectación subcutánea y muscular en un recién
nacido que, por clínica y técnicas de imagen (tomografía
computada y ecografía), fue orientado como una tumoración multifocal,
probablemente una fibromatosis (miofibromatosis) o xantogranulomatosis.
Historia
Clínica.
Recién nacido varón que presentaba
en el período neonatal inmediato múltiples nódulos subcutáneos,
de consistencia firme y adheridos a planos musculares profundos, de 1-3 cm de
diámetro, localizados en cuero cabelludo, cuello, axilas, dorso, extremidades
inferiores y una masa glútea izquierda de mayor tamaño.
Se realizó estudio por ecografía y
eco-doppler (fig.1) de lesiones
superficiales y se practicó una primera biopsia de una lesión
axilar. Se completó el estudio por imagen con una tomografía computada
toraco-abdominal para despistaje de lesiones adicionales (fig.
2). La biopsia de un segundo nódulo adyacente permitió
establecer definitivamente el diagnóstico de la lesión. La evolución
posterior demostró la ausencia de afectación visceral, la involución
completa de la lesión glútea y la regresión paulatina de
las demás lesiones con aparición de focos de calcificación.
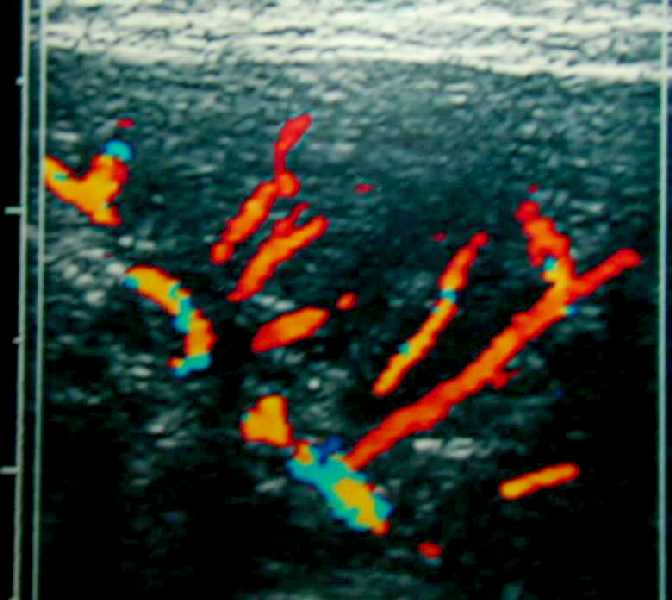
Figura
1 Estudio por ecografía de una de las masas de la región glútea.
(Seleccione imagen para ampliarla).
 |
 |
 |
|
Fig. 1a.
Imagen ecográfica de un nódulo. Es
sólido, complejo, heterogéneo, bien delimitado y con zona
central hipoecogénica.
|
Fig. 1b.
La ecografía Doppler-color muestra flujo vascular intenso alrededor
de las zonas anecoicas de la masa glútea.
|
Fig. 1c. Ecografía
Doppler-color de otro nódulo sólido mostrando aumento difuso
de la vascularización intralesional.
|
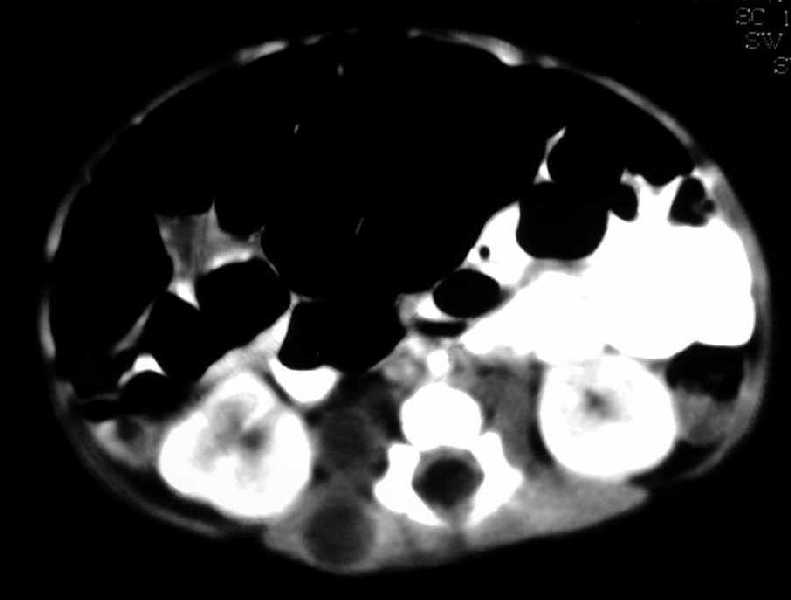
Figura 2 Estudio por tomografía
computada (TC) abdominal y pélvica. (Seleccione
para ver la imagen a 800x600).
 |
 |
 |
|
Fig. 2a.
TC lumbar con contraste; fase arterial. Nódulos
hipodensos con captación periférica del contraste en psoas
y músculos de los canales vertebrales derechos.
|
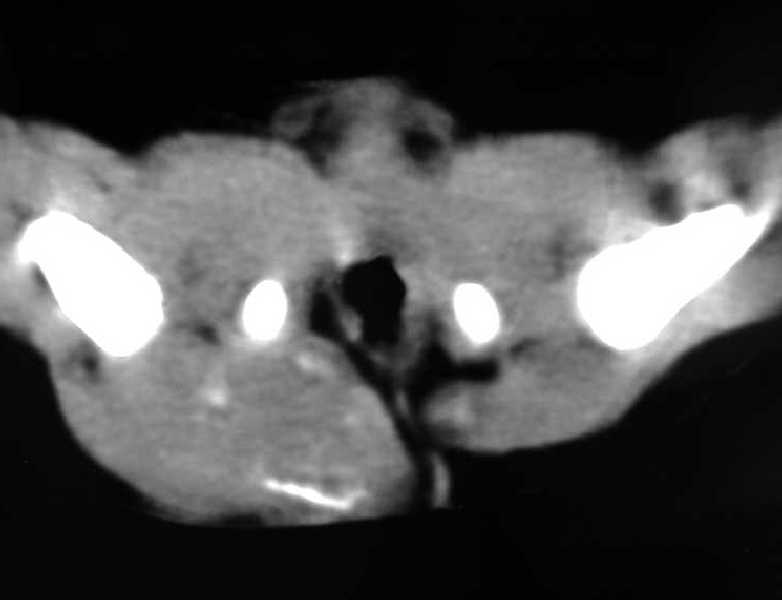
Fig. 2b.
TC pélvica simple. Masa glútea derecha con densidad de partes
blandas, heterogénea y con calcificaciones. Se observa una pequeña
masa hipodensa en el glúteo izquierdo.
|
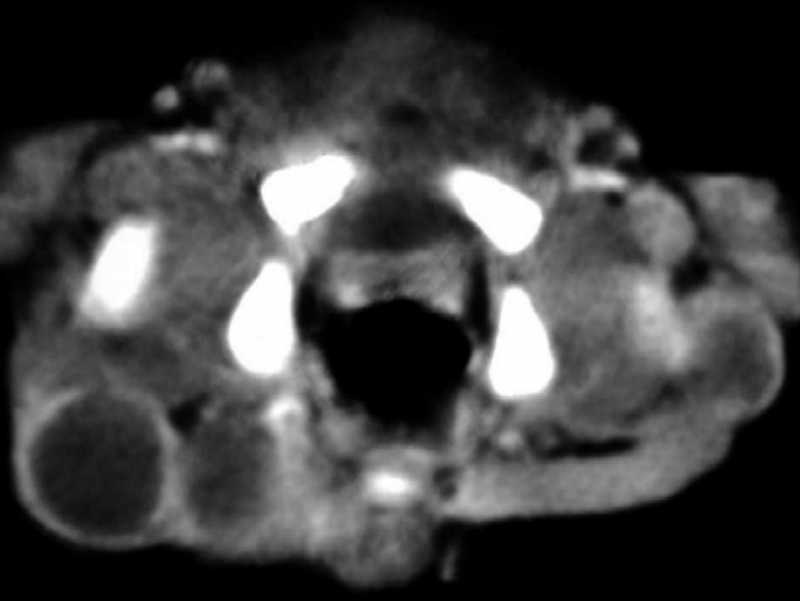
Fig. 2c.
TC pélvica con contraste. Múltiples nódulos hipodensos
con halo de captación periférica del contraste en ambas
regiones glúteas.
|
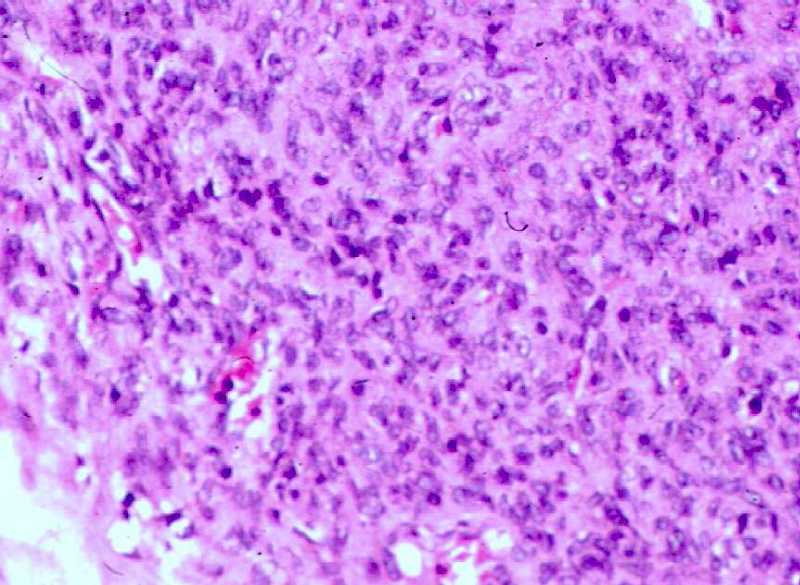
Anatomía Patológica.
La lesión subcutánea biopsiada inicialmente
fue interpretada genéricamente como un tumor mesenquimal con patrón
de hemangiopericitoma, pero no se pudo catalogar definitivamente debido a la
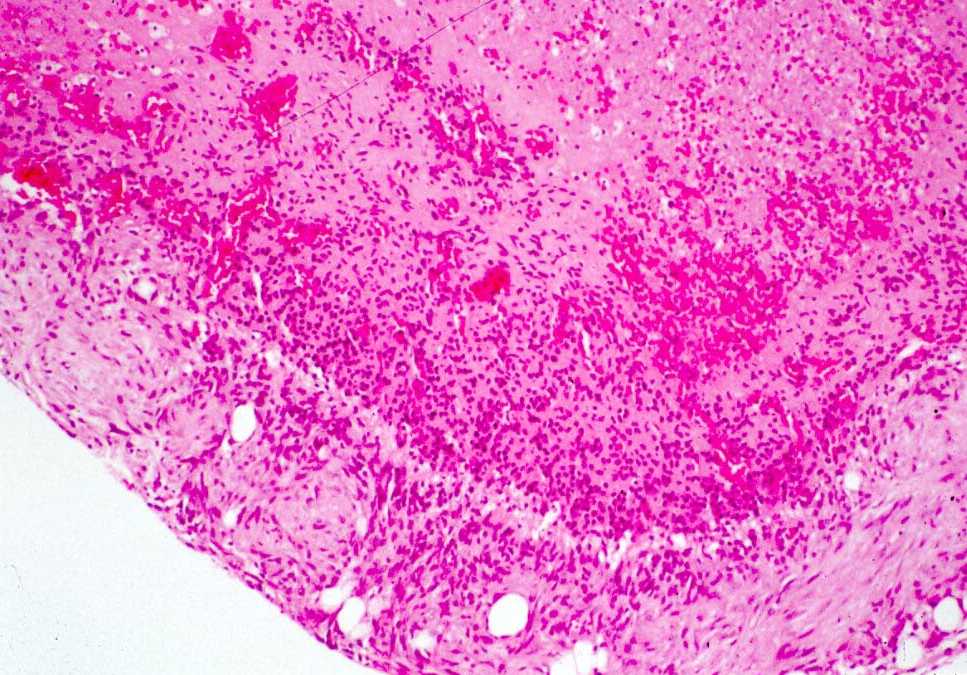
presencia de necrosis extensas (figs. 3a,b).
Figura
3 (a.b). Estudio histológico de la primera biopsia (Seleccione
imagen para ampliarla).
 |
 |
|
Fig. 3a.
Nódulo subcutáneo. Aspecto panorámico mostrando la
periferia del nódulo conservada (abajo) y necrosis central (arriba).
|
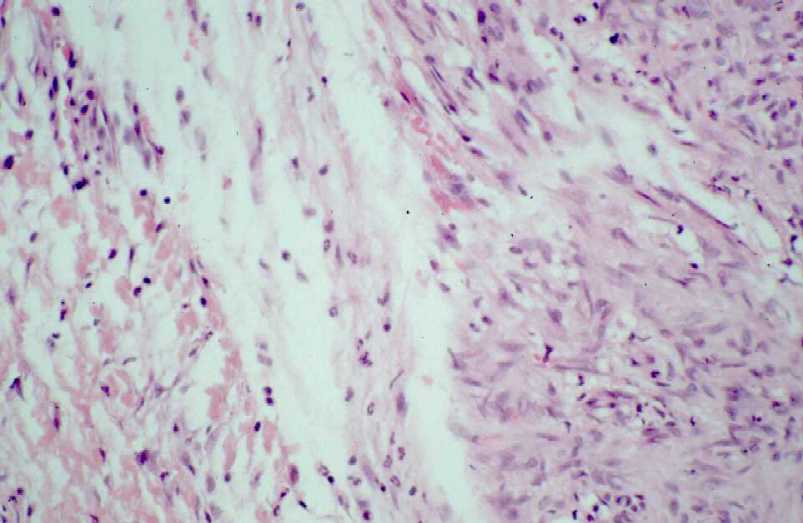
Fig. 3b.
Zona periférica mostrando el área conservada de la lesión,
bien delimitada, con células dispuestas en haces, y de aspecto
miofibroblástico.
|
En la segunda biopsia se obtuvo un nódulo
de 1.5 x 1 cm, denso, de color pardo claro homogéneo al corte. En las
improntas citológicas se obtuvo células redondas, alargadas o
monocitoides, de citoplasma acidófilo y cuyos núcleos presentaban
cromatina nuclear densa. Se observaban focos de células con aspecto fibroblástico.
La imagen, tras fijación en formol, mostraba una proliferación
mesenquimal intramuscular con células uniformes y patrón vascular
focal que sugería un hemangiopericitoma y otras áreas menos densas,
fasciculadas, con células de aspecto fibroblástico. Se observó
la típica distribución zonal, con patrón maduro miofibroblástico
en la periferia (fig. 4a) y patrón vascular central
(fig. 4b). La lesión no infiltraba el músculo
adyacente, del que se hallaba bien delimitada. El estudio inmunohistoquímico
fue negativo para las citoqueratinas, positivo para la vimentina y la actina
de músculo liso, con patrón en nidos perivasculares, negativo
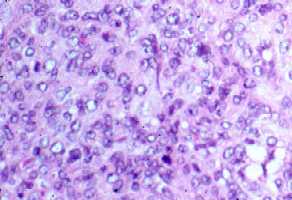
para la desmina, el EMA y el S100, y débilmente positivo para CD68. El
índice mitósico fue de 25/10 campos de gran aumento (figs.
5a,b).
Figura
4 (a.b). Estudio histológico de la segunda biopsia. (Seleccione
imagen para ampliarla).
 |
 |
|
Fig. 4a.
Periferia del nódulo, mostrando un área miofibroblástica
con disposición fascicular esbozada (a la izquierda), adyacente
a tejido más sólido, con marcadas celularidad y eosinofilia.
|
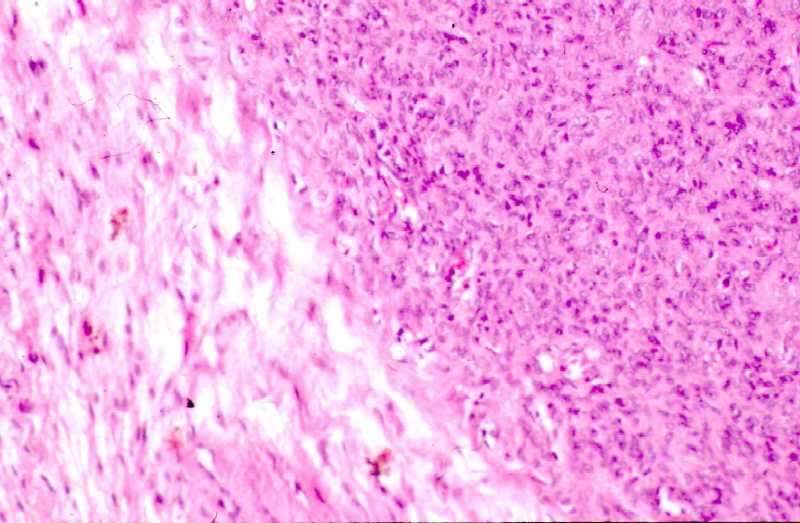
Fig. 4b.
Zona central con patrón vascular sugestivo de hemangiopericitoma.
|
Figura
5 (a,b). Estudio histológico de la segunda biopsia. (Seleccione
imagen para ampliarla).
 |
|
 |
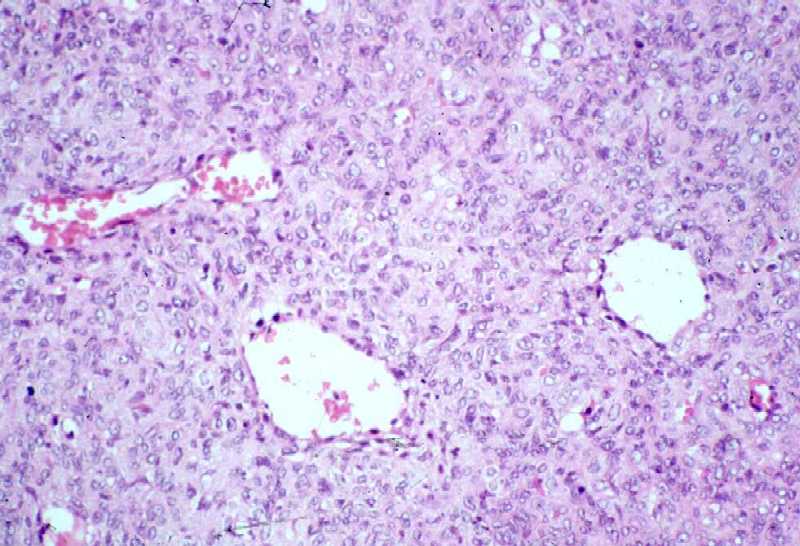
Fig. 5a.
La zona sólida de la fig. 4a muestra células
redondas, monocitoides o alargadas, con eosinofilia y discreto pleomorfismo
(400 aumentos).
|
|
Fig. 5b.
Celularidad menor que en la periferia y componente colágeno con
mitosis (400 aumentos).
|
Discusión.
La miofibromatosis congénita fue descrita
por Stout en 1954 (1). Se trata de una
tumoración generalmente multinodular, aunque más recientemente
según el Instituto de patología de las fuerzas Armadas (AFIP)
son dos veces más frecuentes las formas solitarias (2).
La histogénesis ha sido muy discutida como reflejan la diversidad de
denominaciones que ha recibido. Actualmente se considera el término de
miofibromatosis más adecuado ya que los datos histológico en microscopía
óptica y electrónica son más sugestivos de origen muscular
liso y que, a diferencia de la fibromatosis agresiva, no infiltra musculo esquelético.
En el período neonatal no ofrece dudas con respecto al diagnóstico;
por el contrario, en los primeros años de vida plantea el diagnóstico
diferencial con la neurofibromatosis, que se caracteriza por la presencia de
manchas "café au lait" y la historia familiar, y se diferencia de la
fibromatosis juvenil hialina, entidad rara que suele afectar niños mayores
de 2 años y que cursa con nódulos subcutáneos y deformidad
articular. La presencia de necrosis y mitosis no es sugestiva de malignidad,
un hallazgo que es propio de los tumores de partes blandas infantiles. La miofibromatosis
congénita suele curar tras excisión de los nódulos cutáneos
o músculo-esqueléticos aunque algún tumor, incluso de gran
tamaño puede evolucionar a la regresión espontánea. La
localización visceral asociada, sea esta pulmonar, cardíaca o
gastrointestinal condiciona la clínica y la evolución en los casos
de miofibromatosis congénita generalizada.
a
1.-
Stout. A.P. Juvenile fibromatoses. Cancer 7: 953,1954.
2.-
Enzinger, F.M., Weiss, S.W. En "Soft tissue tumors", Ed. CV. Mosby, U.S.A. Ed
1983, pp.78-83 y 86.
III Congreso Virtual Hispanoamericano de Anatomía Patológica,
Abstract 487
(C) 1999, S. Anatomía patológica, Radiología
y Pediátricas, Vall d'Hebron Hospitals.