OBJETIVOS: Presentación de un caso clínico-patológico de síndrome de Cushing de origen primariamente suprarrenal por hiperplasia cortical nodular masiva, cuadro que da a la glándula un aspecto pseudotumoral susceptible de ser confundido con neoplasia.
MÉTODO: En una revisión de patología quirúrgica de la glándula suprarrenal recientemente realizada en nuestro hospital, se encontraron un total de 82 piezas de adrenalectomía correspondientes al período de veinte años 1978-1998. De estas 82 piezas, 44 (53.6%) fueron casos de patología cortical y 10 de ellas (12.1%), hiperplasias. Un solo caso correspondía a una hiperplasia cortical macronodular con agrandamiento glandular masivo.
CASO CLÍNICO: Varón de 53 años, alcohólico crónico con obesidad cushingoide e hipercortisolismo, que en el estudio radiológico mostró un aumento de tamaño, asimétrico, de ambas glándulas suprarrenales, con presencia de macronódulos. Los estudios analíticos y radiológicos de la silla turca pusieron en evidencia el carácter primario del hipercortisolismo. Se realizó adrenalectomía de la glándula dominante (la izquierda, cuya mayor actividad se demostró en la gammagrafía con I 131). Los niveles de cortisol post-operatorio se normalizaron y se han mantenido hasta la fecha, 18 meses después de la intervención quirúrgica. La glándula extirpada pesaba 104 grs. y presentaba una transformación macronodular total.
CONCLUSIONES: La hiperplasia macronodular con marcado agrandamiento glandular es una rara causa de hipercortisolismo suprarrenal primario autónomo, hipófiso- independiente. Siempre es bilateral. Las glándulas están significativamente aumentadas de peso (de 60 a 180 grs. de peso conjunto) y muestran nódulos que van desde escasos milímetros hasta 4 cms., amarillos o dorados, no encapsulados. Las células que los constituyen son poco activas, por lo que es necesario un gran aumento de tamaño glandular para que produzca el síndrome de Cushing. Los aspectos clínicos y bioquímicos y la radiología pueden ser fuente de confusión y sugerir una neoplasia córtico-suprarrenal. El tratamiento es la adrenalectomía.
hiperplasia córticosuprarrenal; hiperplasia macronodular; síndrome de Cushing primario.
La hiperplasia córtico-suprarrenal adquirida (HCSA), siempre bilateral, puede ser difusa o nodular. La mayor parte de los casos de HCSA difusa (con o sin micronódulos acompañantes) se definen como ACTH-dependientes, sea la fuente de ACTH hipofisaria o ectópica (1). Por el contrario, casi todos los casos de HCSA macronodular son ACTH-independientes (2). Debe tenerse en cuenta que, en ocasiones, la hiperplasia suprarrenal difusa de la enfermedad de Cushing se asocia a algunos nódulos, que suelen ser micronódulos, pero en un 10-20% de los casos son macronódulos; se supone que estos casos representarían un estadio evolutivo avanzado de una hiperplasia difusa en la que la lesión se habría transformado de hipófiso-dependiente en suprarrenal-dependiente (1). En estos casos se observan nódulos que miden entre 0,5 y 5 cms. de diámetro (3), con una media de 16 grs. de peso glandular (4).
Dentro de las HCSA nodulares existen dos variantes morfológicamente distintas: por un lado, la hiperplasia córtico-suprarrenal nodular pigmentada, que forma parte del llamado complejo de Carney (caracterizado por enfermedad de Cushing, pigmentación cutánea, mixomas cutáneos y cardiacos, adenomas hipofisarios secretores de GH y algunos raros tumores, como el "schwanoma melanocítico psamomatoso" o el "tumor de células de Sertoli del testículo con células grandes y calcificaciones") (1); por otro, la hiperplasia macronodular córtico-suprarrenal con agrandamiento importante de la glándula, que siempre es hipófiso-independiente y ACTH-independiente. Esta última variante es francamente infrecuente y, en ella, el peso combinado de la glándulas suele ser de 60 a 200 grs. (4,5). Estas glándulas presentan un agrandamiento muy importante que no se observa en otros cuadros hiperplásicos (5), llegan a simular una neoplasia suprarrenal (3) y están constituidas por grandes nódulos amarillo-dorados, algunos de los cuales alcanzan los 3 ó 4 cms. de diámetro. El promedio de edad de los pacientes es de 50 años, con un ligero predominio masculino, y la duración del síndrome de Cushing suele ser prolongada, de 1 a 10 años (5). A continuación presentamos un caso de esta entidad tratado con adrenalectomía unilateral de la glándula dominante, con remisión sintomática y bioquímica del síndrome de Cushing,
En una revisión de los Archivos del Servicio de Anatomía Patológica del Hospital de Gran Canaria "Dr. Negrín" se contabilizaron un total de 82 piezas de adrenalectomía en 81 pacientes durante el período de Abril de 1978 a Mayo de 1998. La distribución por patologías y porcentajes relativos están representados en la Tabla I.
La patología tumoral supuso un 67% (55 piezas) de la serie, con 7 metástasis extirpadas y 48 tumores primarios (75,6%). Dentro de los tumores primarios hubo 14 originados en la médula suprarrenal y 34 de origen cortical.
Dentro de la patología no neoplásica predominaron las hiperplasias córtico-suprarrenales (10 casos, 37%): 7 de carácter difuso, 2 nodulares y 1 caso único de hiperplasia macronodular con agrandamiento glandular masivo.
Paciente varón de 53 años, fumador, que ingresó para estudio de hipercortisolismo. Presentaba aumento de 15 kgs. de peso en los últimos dos años, sin estrías ni debilidad muscular, e hipertensión arterial, diagnosticada 5 meses antes y no tratada. Entre los antecedentes constaba etilismo crónico con pancreatitis crónica; tres años antes había padecido el último episodio de pancreatitis aguda y se sometió a colecistectomía.
En la exploración física se observó obesidad centrípeta y ginecomastia bilateral de predominio izquierdo. No había visceromegalias, ascitis ni edemas. La tensión arterial era de 170/100 mmHg y la frecuencia cardíaca, de 80 l.p.m. En el estudio analítico destacaban la hipercortisoluria y la hipercortisolemia, que no se suprimían con los tests de supresión con Dexametasona. En sangre se identificó macrocitosis e hipercolesterolemia (270 mgrs./ml), así como aumento de la gammaGT.
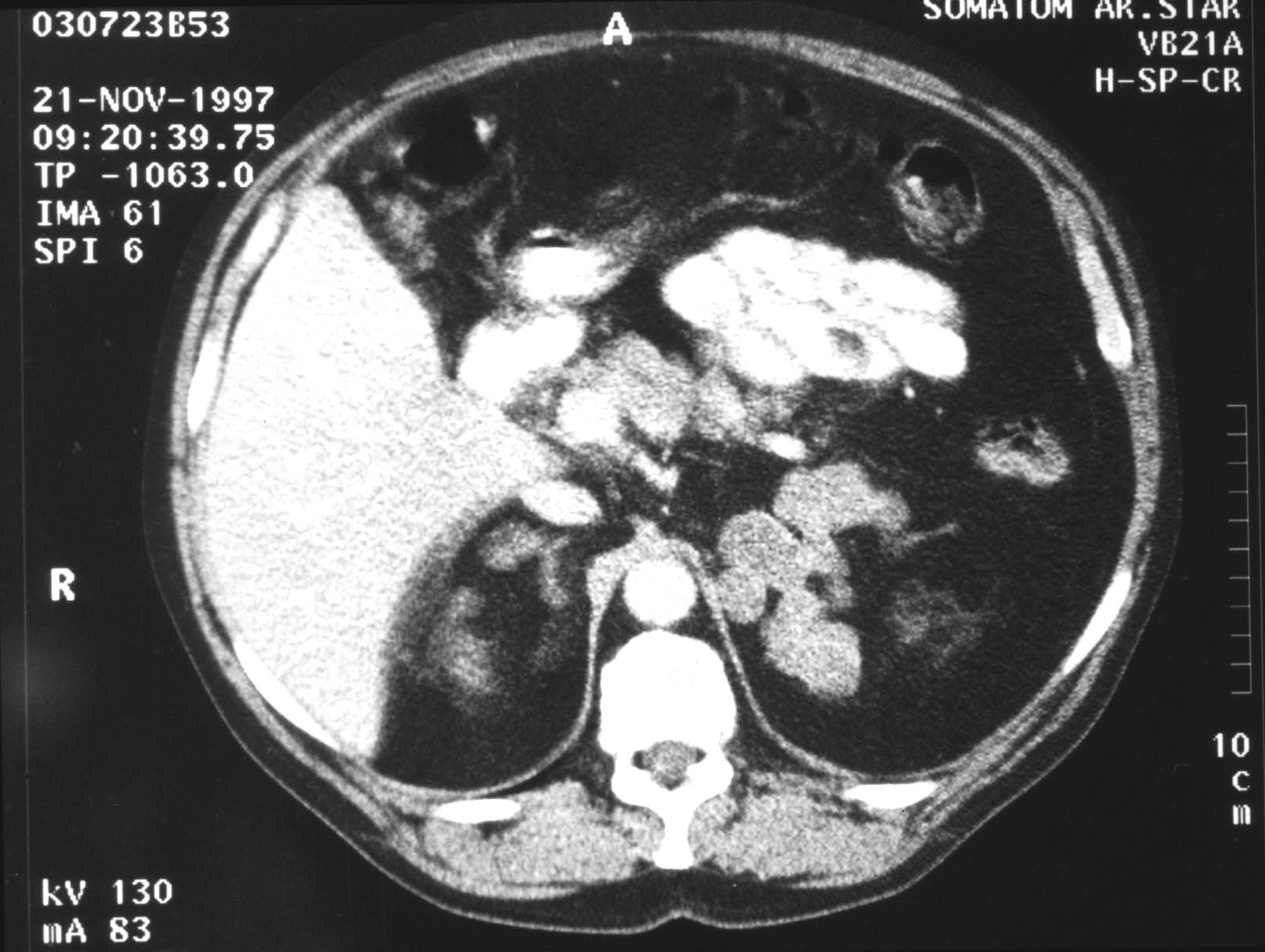
El estudio radiológico incluyó TAC abdominal en la que se ponen de manifiesto glándulas suprarrenales muy aumentadas de tamaño, con múltiples macronódulos que se realzaron discretamente tras administración de contraste (Fig 1). Se efectuó gammagrafía suprarrenal con I 131-Norcolesterol, observándose una actividad bilateral intensa y patológica, más marcada en la suprarrenal izquierda. La cuantificación relativa de la actividad fue del 39,4% en la supararrenal derecha y del 60,6% en la izquierda. El estudio de Resonancia Magnética Nuclear de la silla turca fue normal.
Se realizó adrenalectomía izquierda, cursando el postoperatoprio con hematoma en lecho suprarrenal, que se solucionó espontáneamente, y derrame pleural izquierdo hemático tratado con toracocentesis. Los valores de cortisol postoperatorio fueron, en todo momento, normales.
A los dos meses el paciente reingresó por malestar, astenia, alteraciones digestivas (vómitos, intolerancia a sólidos), y dolor en articulación del hombro. Se detectaron hormonas tiroideas discretamente elevadas, sin hipercortisolismo ni enfermedad de Addison. La gammagrafía tiroidea con Tc99 señaló hipocaptación. La TSH estaba suprimida. A lo largo del ingreso la función tiroidea se fue normalizando y se dio el alta con el diagnóstico de hipertiroidismo inducido por la administración de yodo (en los estudios de imagen, curas con antisépticos yodados, etc.).
El período de seguimiento ha sido de 18 meses. En las últimas semanas el paciente ha notado molestias lumbares atribuibles a una posible eventración post-quirúrgica. Todos los controles clínicos y analíticos han sido normales en este tiempo
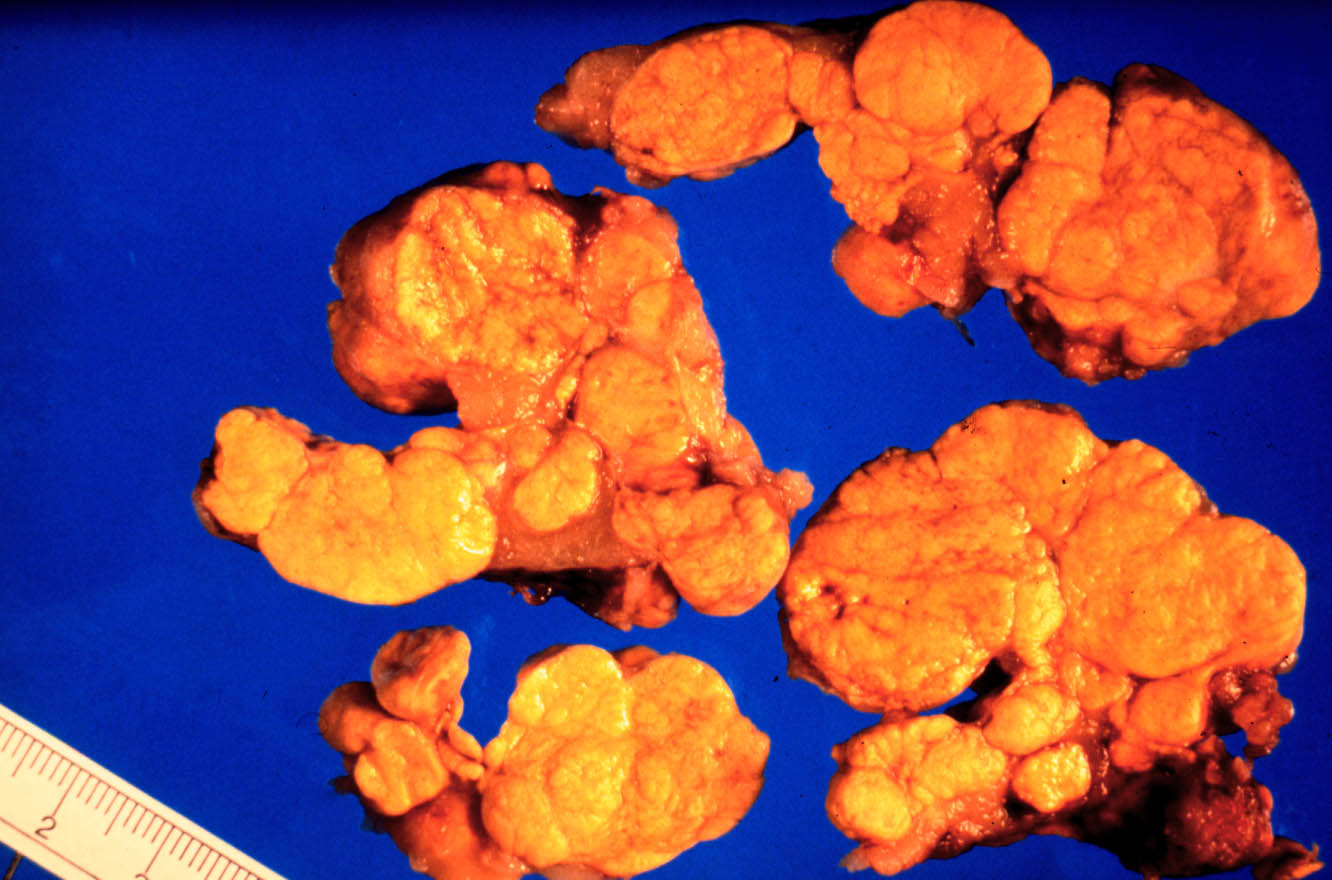
Tras disección cuidadosa y extracción de 58 grs. de grasa retroperitoneal, la suprarrenal izquierda, de 9,8x6x4 cms., pesaba 104 grs. y estaba constituida por múltiples nódulos yuxtapuestos que medían entre escasos milímetros y 3 cms. de diámetro. A la sección todos los nódulos eran similares, sólidos, homogéneos, de un vivo color amarillo-dorado (Fig 2A), sin áreas quísticas, necróticas ni hemorrágicas.
Pese a las múltiples secciones realizadas seriadamente no pudo identificarse macroscópicamente tejido medular.
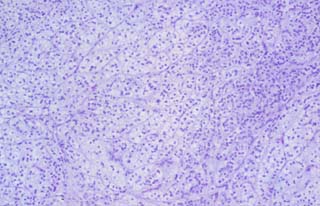
Histológicamente, los nódulos son, asimismo, muy homogéneos, formados por lóbulos y masas de células de amplio citoplasma pálido, mezcladas con pequeños acúmulos de otras células menores, de escaso citoplasma acidófilo y núcleos más oscuros (las llamadas células "compactas" ), sin atipia ni actividad mitótica relevante (Fig. 2B). Los nódulos están, en general, bien definidos, pero no encapsulados, aunque focalmente las células se mezclan libremente con la grasa vecina.También se observan adipocitos en el centro de algunos nódulos, así como regueros de células linfoides entre las células corticales hiperplásicas y un foco de calcificación distrófica sobre necrosis isquémica intranodular. En varias secciones histológicas se detectan ribetes de tejido medular suprarrenal normal, comprimido entre los nódulos, con pequeños focos de córtex atrófico.
La frecuencia de presentación de la hiperplasia macronodular córtico-suprarrenal con agrandamiento masivo glandular (HMCAM) es muy baja. Solo se han publicado 23 casos en la literatura anglosajona hasta 1995 (4) y una docena más hasta 1998, la mayoría de origen japonés (2,6-9). Las series son cortas y la mayor, la de Sasano y cols. (2), registra 6 casos intervenidos.
Clínicamente se manifiesta con un síndrome de Cushing de evolución variable (4), que en varios casos era un síndrome de Cushing preclínico (8,10) en los que el descubrimiento de masas tumorales suprarrenales se debió a estudios radiológicos por otra causa ("incidentalomas"). En esos casos los pacientes no presentaba síntomas todavía y el cortisol sérico era normal, si bien ya existía pérdida del ritmo diurno de secreción cortisólica, ausencia de supresión con Dexametasona y ACTH indetectable (10). En uno de estos casos los mineralocorticoides eran normales preoperatoriamente y disminuyeron tras una adrenalectomía subtotal, lo que llevó a Yamada y cols. (10) a suponer que en la HMCAM puede haber secreción simultánea de gluco y mineralocorticoides. En un único caso se describe la asociación de síndrome de Cushing con feminización (3); aunque en el aquí expuesto existían ginecomastia bilateral e impotencia, creemos que estaban más en relación con su etilismo crónico.
Los estudios endocrinológicos muestran, en la HMCAM, elevados niveles de cortisol basal junto con pérdida del ritmo circadiano y ausencia de respuesta a la supresión con Dexametasona. La ACTH en plasma es indetectable y la silla turca, normal. Se ha descrito un aumento del cortisol plasmático en respuesta a la hipoglucemia inducida por insulina, lo que sugiere unas glándulas suprarrenales hiperreactivas a pequeños cambios de la ACTH, no detectables por radioinmunoensayo, motivados por la insulina u otros factores desconocidos (3). En un trabajo (9) se describen aumentos estadísticamente significativos del cortisol tras inyección de Lisina-vasopresina, tanto en vivo como "in vitro", de manera similar a lo que ocurre en los pacientes con adenoma productor de cortisol.
Macroscópicamente, las glándulas sufren una transformación multinodular bilateral que aumenta su peso entre 10 y 30 veces, con nódulos que miden hasta 4 cms. de diámetro (5), sin que se reconozca tejido suprarrenal normal entre ellos. En el estudio histológico existe proliferación de dos tipos celulares: células de amplio citoplasma claro y células de citoplasma más escaso y acidófilo, de tipo compacto (3), con mezclas variables entre ellas. Puede ser difícil descubrir el tejido medular normal en secciones tomadas al azar; en nuestro caso sí lo observamos tras una adecuada orientación de la pieza. La existencia de atrofia del córtex internodular se describe en dos artículos (7,9) de forma semejante a lo que ocurrió en nuestro caso; este hallazgo sugiere una afección ACTH-independiente (3). Igualmente, los nódulos pueden estar mal definidos, con extensión de células corticales entre las células grasas, y en ocasiones aparecen metaplasia lipomatosa o pequeños focos de necrosis y calcificación isquémica (3), hallazgos todos identificados en el estudio microscópico de nuestro caso.
Varios grupos de autores japoneses han caracterizado, mediante técnicas inmunohistoquímicas, el patrón enzimático relacionado con la esteroidogénesis de las células de los macronódulos. Así, observaron inmunorreactividad para p450c17, principalmente en las células pequeñas compactas (2,6,8), lo que se confirmó en algunos casos mediante hibridación "in situ" (2). Los enzimas p450scc, p450c21 y p450c11 se expresaron igualmente en las células "compactas" (2), mientras que la 3beta-hidroxiesteroidedeshidrogenasa es expresada exclusivamente por las células grandes y claras (2,6,8). La expresión enzimática es mucho menos intensa que en los casos de hipercortisolismo relacionado con adenoma suprarrenal (7) o en la hiperplasia nodular cótico-suprarrenal pigmentada (2). Todo lo antedicho implica, por un lado, que la HMCAM tiene un patrón emzimático peculiar y exclusivo (2) y, por otro, indica una esteroidogénesis poco eficaz que podría contribuir a la relativamente baja producción de cortisol (2). Así se explicaría que las glándulas alcancen tan grandes tamaños antes de producir un síndrome de Cushing (8,10) y que éste sea de aparición tardía (más de 10 años de diferencia en relación con la enfermedad de Cushing clásica ACTH-dependiente) (2,3) y no siempre bien desarrrollado, como ocurrió en el caso que presentamos.
El diagnóstico diferencial de la HNCAM debe hacerse con las siguientes entidades :
1.- Neoplasia. La confusión surge ante el gran aumento de tamaño, frecuentemente asimétrico, de las glándulas, que se evidencia con los modernos estudios radiológicos. No obstante, la bilateralidad y la multinodularidad son la regla, si bien podrían inducir, en algunos casos, a sospechar metàstasis.
2.- Estadio avanzado de una hiperplasia difusa con enfermedad de Cushing y formación de macronódulos. Los nódulos por lo general miden menos de 2 cms., pero se han descrito de hasta 5 cms. El problema diagnóstico se agravaría en casos de paso a una fase de menor sensibilidad a la ACTH o incluso de independencia de la ACTH (hipercortisolismo" terciario") (3). En estos casos, el peso combinado de ambas glándulas suele ser notablemente inferior al de la HMCAM; sin embargo no existen límites definidos que separen o relacionen definitivamente la hiperplasia macronodular sobre hiperplasia difusa y la HMCAM (3).
3.- Hiperplasia (también llamada "displasia") córtico-suprarrenal nodular pigmentada, rara afección asociada a hipercortisolismo ACTH-independiente. Esta entidad se observa como un componente del llamado Complejo de Carney, hereditario, de transmisión autosómica dominante y que incluye mixomas cardíacos y cutáneos, pigmentación cutánea, fibroadenomas mixoides de mama y tumores testiculares, entre otras posibles anomalías (4). En esta hiperplasia el tamaño de los nódulos es variable, aunque, en general, menor que en la HMCAM y los nódulos están constituidos histológicamente por células eosinófilas similares a las de la zona reticular del córtex, con abundante pigmento (3).
4.- Suprarrenales con nódulos incidentales sin significación clínica. Suelen ser múltiples, aumentan de número con la edad y se identifican en un 3% de las autopsias (1) y entre el 0,6 y el 1,3% de pacientes asintomáticos (4). Un ejemplo especial es el de los nódulos pigmentados corticales incidentales, localizados en la zona reticular, a veces múltiples y/o bilaterales, y que contienen pigmento de tipo lipofuscínico o neuromelanina (4). Mucho más raro es el caso de los adenomas multiples funcionantes, que pueden coexistir con nódulos no funcionantes (4).
La mayoría de los autores coinciden en que el tratamiento de elección de la HMCAM es la adrenalectomía bilateral (4,5,7). No obstante, el bajo nivel del hipercortisolismo en casos concretos (como en el aquí presentado) permitiría la adrenalectomía subtotal (10) o unilateral de la glándula dominante, con seguimiento cuidadoso del paciente a fin de completar el tratamiento quirúrgico en el momento en que fuese necesario.
TABLA 2

BIBLIOGRAFÍA